
13: Síntesis de Vitamina B₂₂




- Última actualización
- 30 oct 2022
- Guardar como PDF
( \newcommand{\kernel}{\mathrm{null}\,}\)
La síntesis total de Vitamina B 12 se logró en 1973 mediante una gran colaboración entre el grupo de R. B. Woodward en la Universidad de Harvard (EE.UU.) y el grupo de A. Eschenmoser, Instituto Federal Suizo de Tecnología (ETH), Zúrich, Suiza. Tomó cerca de doce años y más de dos docenas de científicos de alto nivel para completar esta gigantesca tarea. El logro es elogiado de diversas formas por químicos orgánicos: logro monumental en los anales de la química orgánica sintética; un avance; una piedra de una milla en síntesis orgánica; inigualable incluso después de 40 años; una tarea no menos aventurera que concurrente en el Monte Everest. En el momento en que comenzó la aventura fue el reto más formidable en química sintética, que pocos se habrían atrevido. El anuncio de su síntesis marcó la mayoría de edad de la química orgánica sintética. Woodward y Eschenmoser trabajaron en estrecha colaboración y competencia durante este ritmo histórico. En el proceso, lograron no sólo una síntesis asombrosa, sino que también abrieron varios campos nuevos para futuras investigaciones. Woodward Hoffmann Rule es la más famosa de las ramificaciones. Al estudiar esta síntesis, el estudiante también debe reflexionar sobre la minuciosidad en su planeación de todos los aspectos del esquema e iniciación de estudios básicos adecuados con mucha antelación para facilitar el esquema principal en las coyunturas apropiadas. Esta larga introducción está justo en sintonía con la duración del esquema, el tiempo tomado y los magníficos logros.
Retroanálisis de laB12 síntesis de vitaminas
La Figura 13.1 muestra la estructura de Vit B 1 2 y las principales características estructurales/desafíos de la molécula compleja.
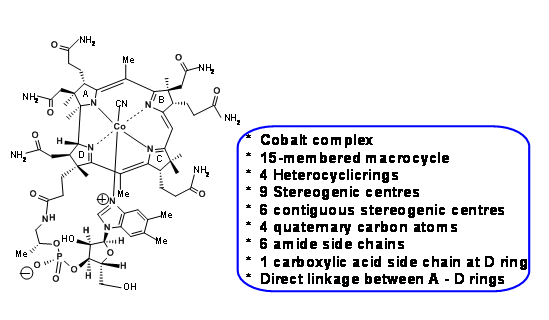
En su estilo inimitable, R. B. Woodward llama la atención sobre estos desafíos al inicio de sus conferencias y al hecho de que tardaron cerca de 50 años en establecer la estructura de Vit B 1 2 por degradación química y finalmente por estudios de difracción de rayos X de Dorothy Hodgkin en 1956. Cabe señalar que la mayoría de los estudios de degradación química, por notables que sean, ocurrieron durante los primeros años del siglo XX, cuando la química orgánica moderna estaba en su elaboración. Los datos espectroscópicos, si los hubiera, eran visibles por su ausencia y la mayor parte de la degradación y la química sintética eran demasiado duras para los estándares modernos (es decir, de los años 60) para esta delicada molécula. De ahí que se tratara de un escenario desalentador. Esto significó que una gran cantidad de química sintética tuvo que ser reinventada para adaptarse a estos desafíos. Dichos aspectos se planificaron a fondo y se pusieron en marcha planes de contingencia con mucha anticipación. Los estudiantes exigentes pudieron vislumbrar esta planeación en esta breve presentación. Las ideas sobre el retroanálisis, como entendemos hoy en día, se encontraban en las etapas de desarrollo durante los años sesenta. Los retroanálisis que utilizamos aquí son adiciones posteriores de otros científicos, basados en los hechos (conferencias, ponencias, etc.) publicados por los dos grupos. Todas esas citas se incluyen al final de la redacción.
La diana sintética se identificó como Ácido Cobírico, debido a que este compuesto era un producto natural y había sido convertido en Vit B 1 2 por Bernhauer K., et.al., (1960) (Figura 13.2). De ahí que la síntesis total de Ácido Cobírico equivaldría a una síntesis formal de Vit B 1 2.
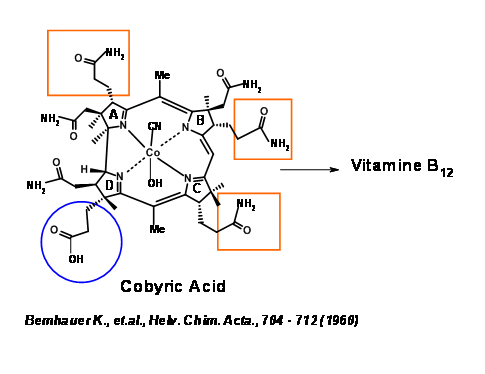
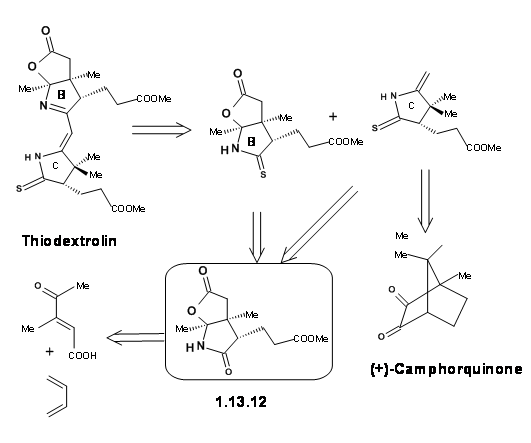
El ácido cobirico tenía siete cadenas laterales de ácido carboxílico, de las cuales cuatro eran restos de ácido propiónico, uno en cada anillo heterocíclico. El principal desafío fue diferenciar la cadena de ácido propiónico en el anillo D de los otros restos de ácido acético y ácido propiónico. Por lo tanto, se decidió que este resto ácido impar estaría enmascarado como nitrilo (Figura 13.3). Eso todavía deja una tarea desalentadora de diferenciación, que podríamos abordar más adelante. Primero se decidió ver la molécula como compuesta por dos mitades: el lado este y el lado oeste. La primera desconexión fue en la unión del anillo A/B en el puente metileno (1.13.3A). La escisión del segundo puente en las uniones del anillo C/D dio a la mitad oriental como tiodextrolina a cargo del grupo de Eschenmoser en Zurich y la mitad occidental como cianobromida a cargo del grupo de Woodward en Harvard (US).
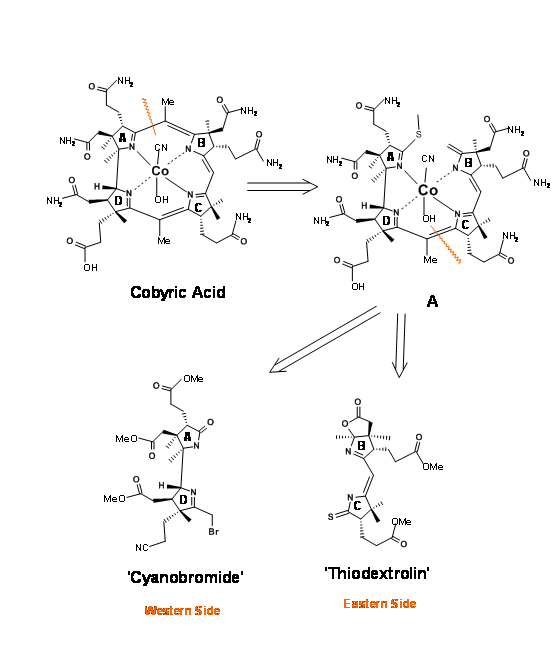
Retroanálisis de cianobromida
Esta mitad occidental tiene una formidable matriz de seis estereocentros contiguos en un marco de ocho carbonos. Obsérvese que los estereocentros se planificaron sobre la base de estereoselectividades conocidas y los anillos de seis miembros se construyeron para proporcionar las cadenas de ácido propiónico (Figura 13.4). El nitrógeno para el anillo A provino de un indol, cuyo anillo de benceno dio las cadenas laterales para el anillo A. El nitrógeno del anillo D vino a través de un reordenamiento de Beckmann. Corrnorsterone (1.13.4A) fue el intermedio clave (piedra angular) que sostenía todos los estereocentros y cadenas en la Mitad Occidental.
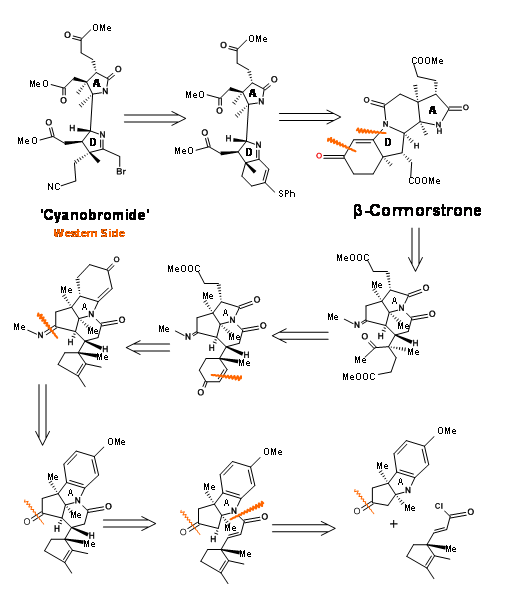
Síntesis de la mitad occidental
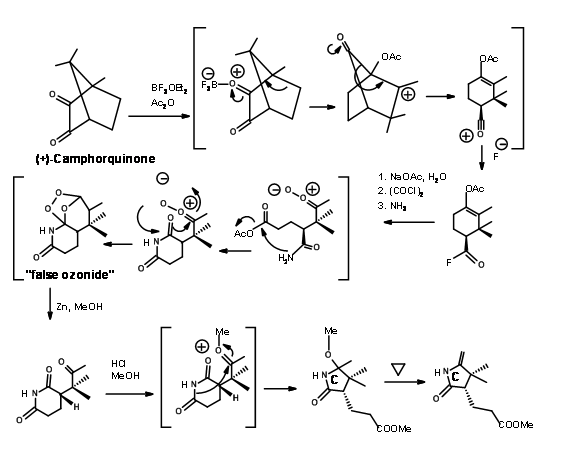
La unidad enantiopura requerida de 1,2,3-trimetilciclopenteno provino de canforquinona como se muestra en la Figura 13.5.
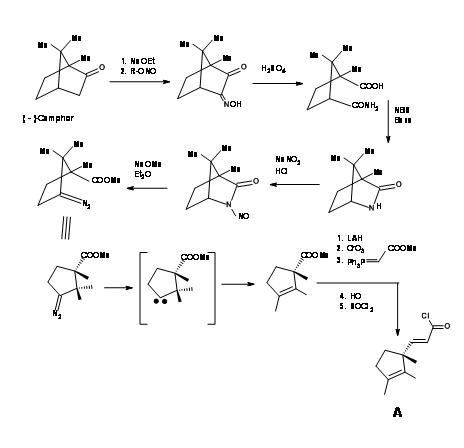
A través de otra síntesis convergente, se fusionó un anillo de cinco miembros con indol en el enlace C2-C3 y se resolvió como se muestra en la Figura 13.6.
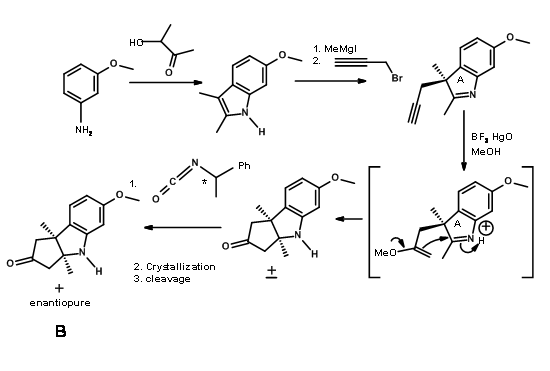
El enantiómero (+) - fue realmente necesario para la síntesis de la diana. El inútil (-) - enentiómero se utilizó como compuesto modelo (para esto fue “casi el único tipo de estudio modelo que consideramos totalmente confiable” — RBW).
Los fragmentos A y B se combinaron y luego se procesaron a Corrnorsterona como se muestra en la Figura 13.7.
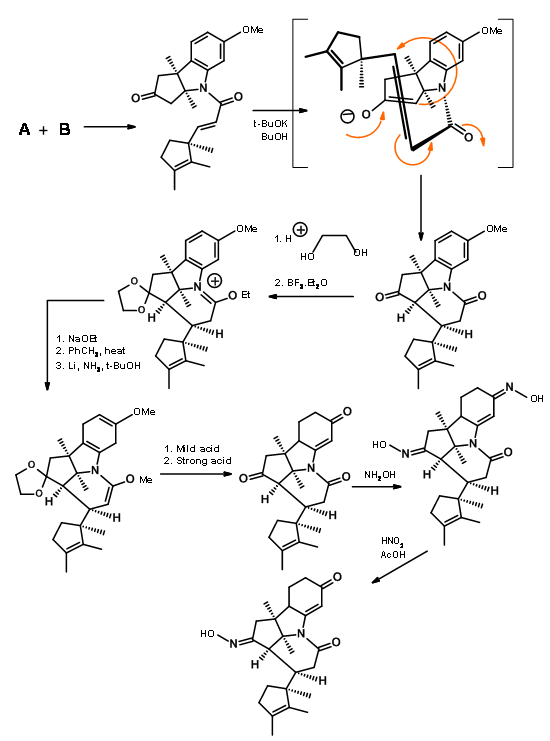
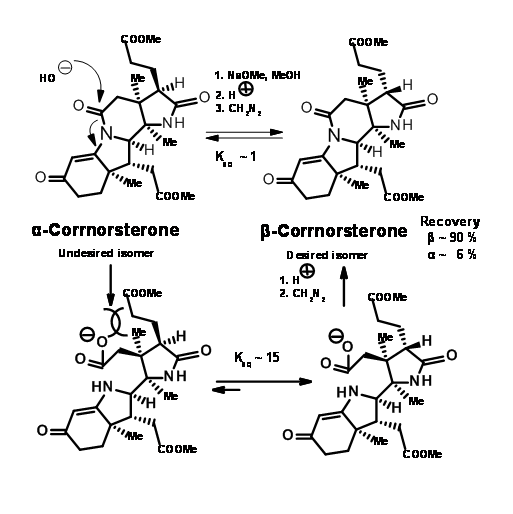
Tenga en cuenta que el proceso de expansión del anillo Beckmann puso en marcha una cascada de reacción, lo que condujo a una condensación de Claisen y la formación del anillo D, todo en un solo paso. También se escindió el carbonilo de imida de seis miembros y se colocó una cadena de acetato en el anillo D. A pesar de esta elaborada planeación y ejecución, el proceso arrojó una mezcla de epímeros en la cadena lateral del ácido propiónico en el anillo A, siendo el isómero requerido un componente menor de la mezcla. El principal producto no deseado no se escinde en el enlace amida debido a la compresión estérica desfavorable en las cadenas laterales en desarrollo (Figura 13.8).
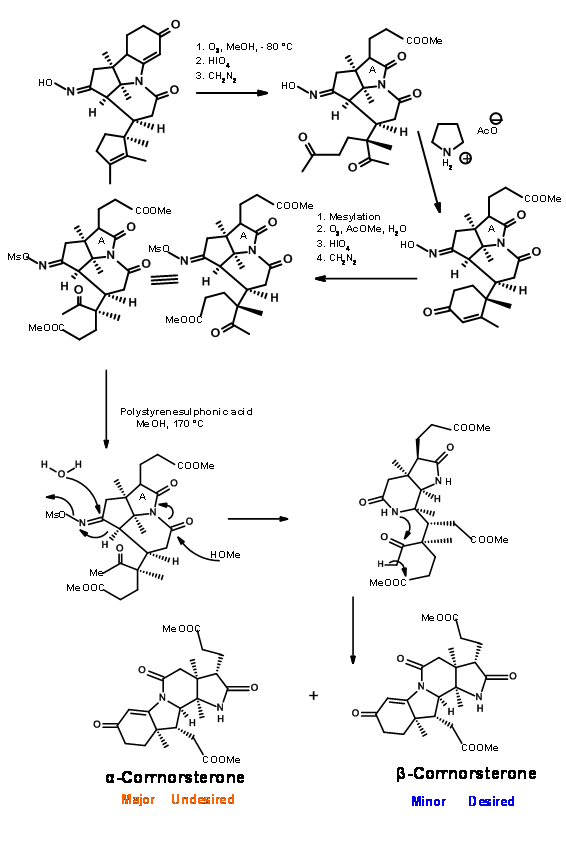
Sin embargo, este desfavorable problema estérico se resolvió pronto. En la hidrólisis bajo condiciones de base fuerte, el anillo de amida se abrió y la cadena lateral del ácido propiónico se isomerizó al isómero menos tenso. Esta podría ser acidificada y esterificada a β-corrnorsterona, con una recuperación del 90% del isómero deseado (Figura 13.9).
Este isómero correcto se trató con una mezcla de metanol y tiofenol en condiciones ácidas (Figura 13.10). Esto puso en marcha dos procesos. El tiofenol atacó a la cetona y activó este centro, mientras que el oxígeno metanol atacó el enlace amida dando lugar a un éster y un tioenol éter.
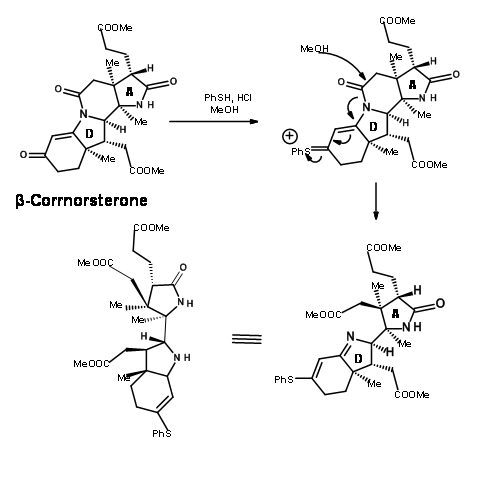
La ozonólisis del tioenol éter a — 90°C escindió la unidad de olefina al compuesto aldehído-tioéster (Figura 13.11). Aquí evolucionó una nueva química interesante. Mientras que los tioésteres son menos reactivos a la hidrólisis ácida y mostraron una reactividad comparable con los nucleófilos de oxígeno, los nucleófilos de nitrógeno fueron únicos. El tioéster reaccionó mucho más rápido que los oxiésteres normales para dar amidas. Así, el tioéster se escindió exclusivamente a amida con amoníaco, dejando tres ésteres metílicos intactos. Luego, el resto aldehído se convirtió selectivamente en alcohol y luego se mesiló en condiciones mixtas de anhídrido/piridina. Esta secuencia también convirtió la fracción amida en nitrilo. El mesilato se convirtió luego en bromuro para dar el intermedio clave cianobromuro.
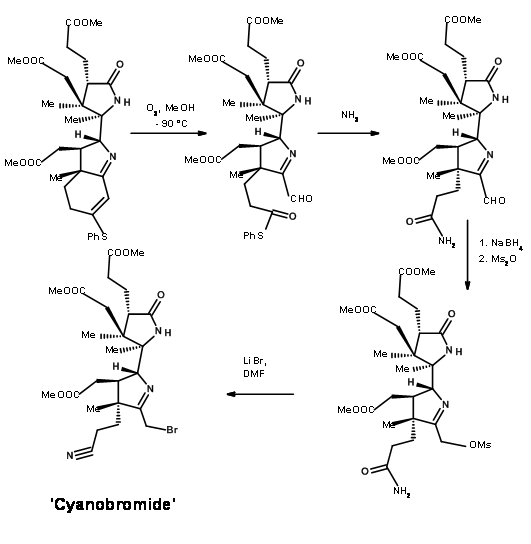
El grupo de Zurich estaba trabajando simultáneamente en la síntesis de la Mitad Oriental llamada Tiodextrolin. Los fragmentos para anillos B y C se planearon vía., un solo intermedio (1.13.12).
La síntesis se inició con una reacción de Diels-Alder para asegurar adecuadamente dos centros asimétricos y se resolvió el racemato. El enantiómero puro se siguió a través del esquema para obtener el segmento del anillo B. El mismo intermedio produjo también el fragmento del anillo C (Figura 13.13).
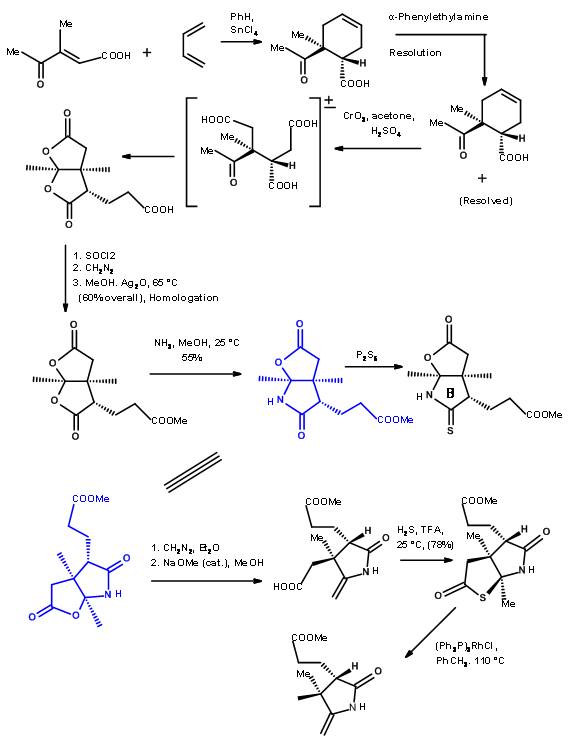
Para conectar dos de estos fragmentos, Eschenmoser había desarrollado dos procedimientos de contracción de sulfuro. A continuación se muestran los mecanismos de los procesos.
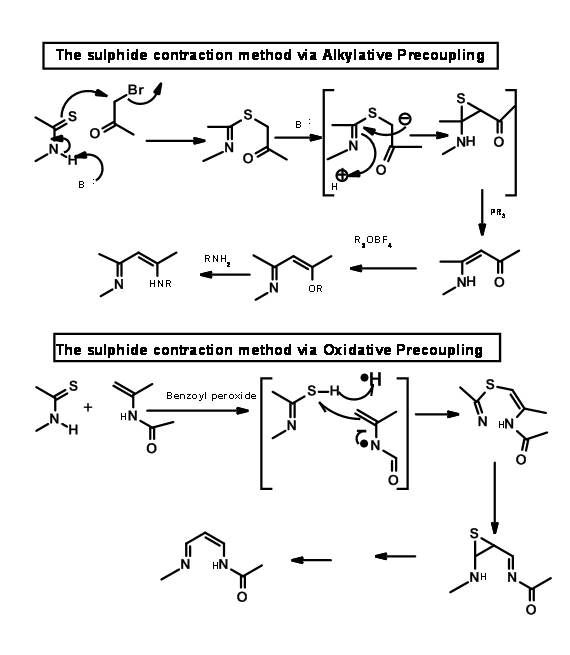
Mediante el procedimiento de acoplamiento oxidativo/extrusión de azufre, acoplaron los anillos B y C como se muestra en la Figura 13.15.
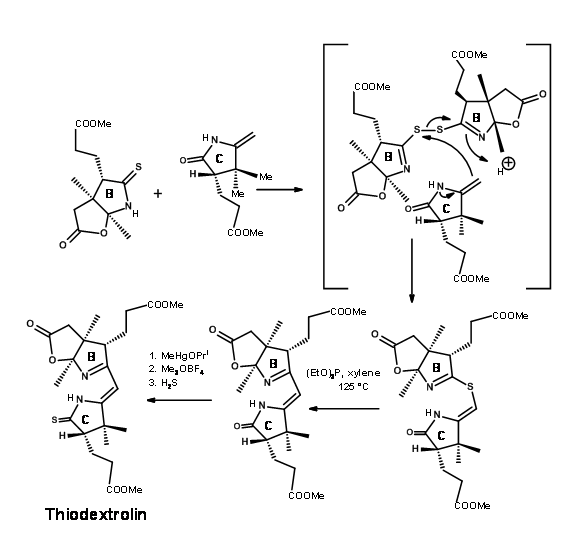
El grupo de Woodward también desarrolló una nueva síntesis para el anillo C a partir de (+) -canforquinona. El esquema se muestra en la Figura 13.16.
Aunque en todos los puntos se había ahorrado mucho cuidado por los estereocentros, no se pudo evitar el problema de los estereoisómeros. La tiodextrolina cristalina que se sintetizó fue en realidad una mezcla de dos estereoisómeros en el resto ácido propiónico del anillo B. Aunque habían purificado la mezcla en este punto, no tenía ningún valor debido a que este estereocentro se debió a nuevas perturbaciones en etapas posteriores. La mezcla se adelantó para el primer acoplamiento en el puente C/ D. Después de un esfuerzo considerable que duró más de un año, se acoplaron primero en el extremo Sur mediante el procedimiento de alquilación/extrusión (Figura 13.17). Obsérvese que el primer producto de alquilación fue un tioéter Tipo I, el cual se isomerizó fácilmente a tioéter Tipo II. El producto se denominó Cianocorrigenolide. Esta isomerización perturbó el estereocentro en el anillo C.
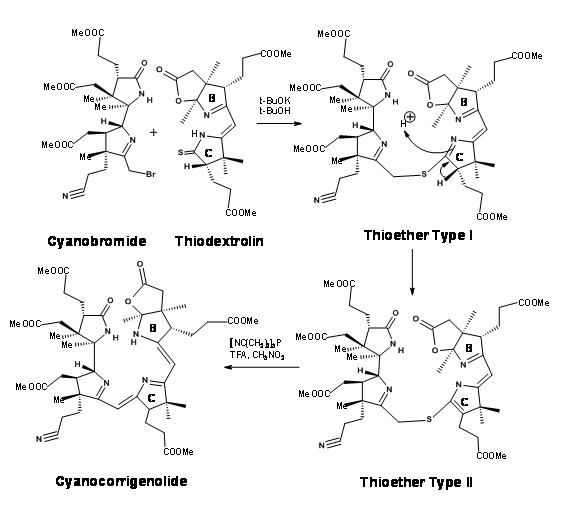
La siguiente fase fue la formación del puente A/B. El compuesto acoplado C/ D se trató primero con pentasulfuros de fósforo seguido de fluoroborato de trimetiloxonio (M e 3 OB F 4) (Figura 13.18). Este procedimiento reemplazó los oxígenos en los anillos A y B por azufre y finalmente al derivado S-metilo. La dimetilamina en metnol escindió el anillo de tiolactona selectivamente a una cadena de dimetilacetamida y una olefina terminal. La olefina era bastante inestable. Esto iba a convertirse inmediatamente en el complejo de cobalto. Este procedimiento no fue fácil. Bajo varias condiciones, el ion cobalto metálico catalizó otras reacciones que condujeron a una extensa “destrucción” del compuesto. Después de varios experimentos se observó que el cloruro o yoduro de cobalto en THF fue único para la cobaltación suave. El proceso de complejación acercó a los anillos A y B. Una reacción catalizada por base permitió la formación del puente y la eliminación del resto azufre, siendo la mejor condición la ciclación catalizada por DBN.
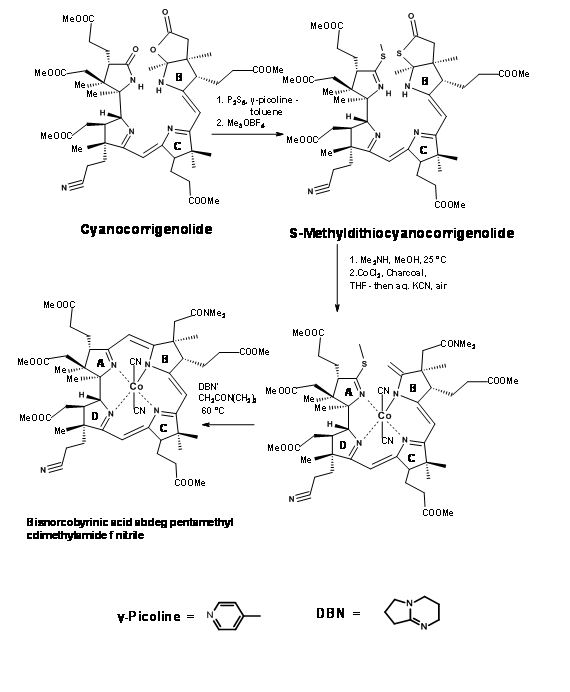
Obsérvese que la transformación global interfirió con el centro asimétrico en el anillo C. No obstante, el puente A/B quedó finalmente en su lugar.
Al grupo de Zurich también se le ocurrió un procedimiento alternativo del complejo Zn en la misma línea. Todas estas manipulaciones fueron ciertamente duras para las (tres) cadenas de ácido propiónico epimerisables en los anillos A, B y C. El producto final se purificó por TLC (“cromatografía en placa”) y se analizó críticamente por HPLC (una nueva herramienta cromatográfica en ese momento). Los cromóforos UV en todos los productos fueron de gran ayuda en esta cromatografía.
La síntesis tiene ahora dos grandes hitos por superar. Hay tres puentes metino activos en la molécula. El puente en los anillos B/C tuvo que ser 'protegido' de la metilación. Esto se logró mediante una reacción de formación de lactona oxidativa en el anillo B (I2, AcOH) (Figura 13.19). Este nuevo centro cuaternario y el carbono cuaternario existente en C12 juntos ejercieron una congestión estérica alrededor de C10. El éter clorometílico ingresó exclusivamente a los centros C5 y C15. La hidrogenólisis de Ra-Ni escindió los tioéteres y el anillo de lactona en un solo paso. A este paso le siguió la esterificación.
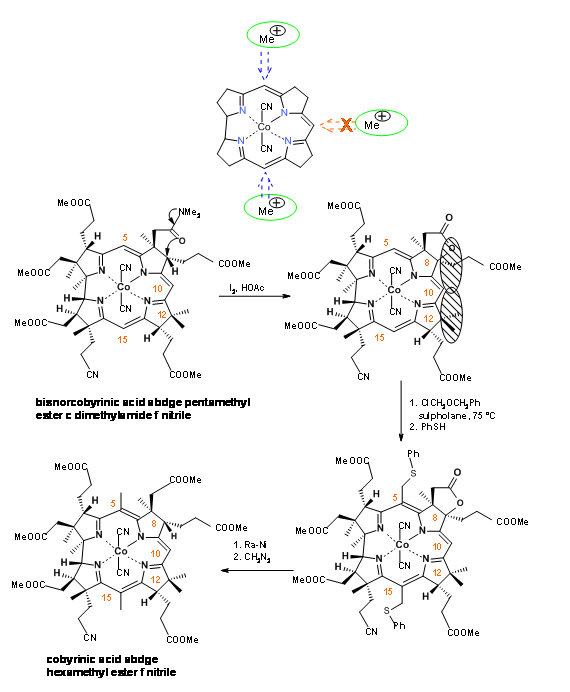
El ácido sulfúrico concentrado convirtió el nitrilo en amida (Figura 13.20) .En esta etapa, se enfrentaron a la difícil tarea de escisión selectiva de amida, en presencia de seis ésteres en la molécula. Después de extensas experimentaciones paralelas, el grupo Harvard redescubrió una eficiente escisión selectiva del resto amida en reactivo N 2 O 2.
El grupo Zurich también salió con un “esquema de corte diabólicamente” para la hidrólisis selectiva del grupo amida en presencia de grupos éster. Este esquema se muestra en la Figura 13.21. Sin embargo, el primero fue el preferido debido a su simplicidad y mejores rendimientos. Sin embargo, la solución de Eschenmoser es testimonio del ingenio humano.
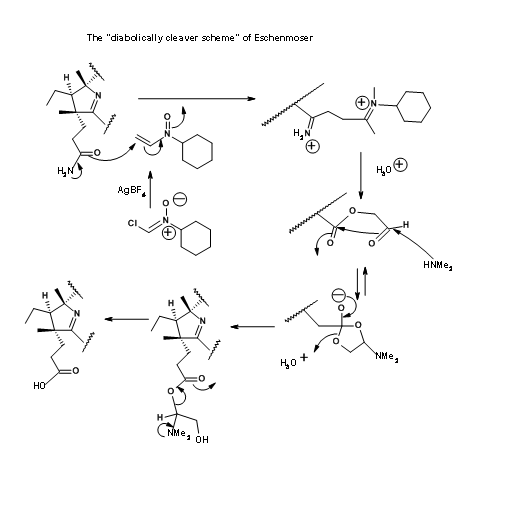
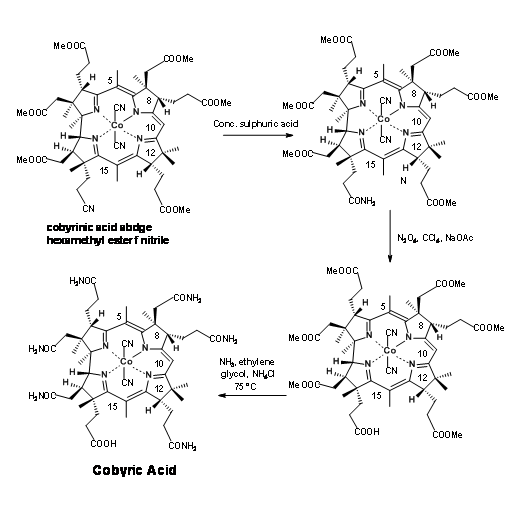
El último paso de esta larga síntesis planteó un problema importante que necesita mención especial. La amidación de ésteres con amoníaco fue el único paso restante para el asalto final a la síntesis de ácido Cobyric. Una mirada más cercana revela que la tarea puede no ser tan fácil. Los restos éster (en particular las unidades de acetato) estaban en ambientes crowed. Paralelamente a estos desarrollos, se estaban llevando a cabo estudios de modelos sobre moléculas muy similares. Con base en estos estudios, cuando el ácido hexametiléster 1 se trató con amoníaco en etilenglicol a 75°C durante 30 horas, el producto obtenido no fue ácido cobyrico sino un ácido pseudocobyrico, cuya estructura se estableció como ácido deshidrocóbirico. Este producto se pudo purificar solo por HPLC. Este fue uno de los productos obtenidos por trabajadores anteriores. Pero no tenían idea de tal complicación. Este misterio tomó varios estudios críticos para resolver. A lo largo de todos estos estudios, se tuvo mucho cuidado para ver que los solventes estaban bien desoxigenados antes de su uso. Se evitó estrictamente el oxígeno de la atmósfera de reacción. La fuente de esta ciclación oxidativa se atribuyó al cobalto, el cual se sospechó que se enlazaba oxidativamente a la posición C9, facilitando la ciclación del anión amida. Esta reacción no deseada tardó mucho en resolverse. Varios estudios estuvieron dirigidos al amoniálisis en condiciones reductoras y también a abrir el anillo lactámico en condiciones de reducción.
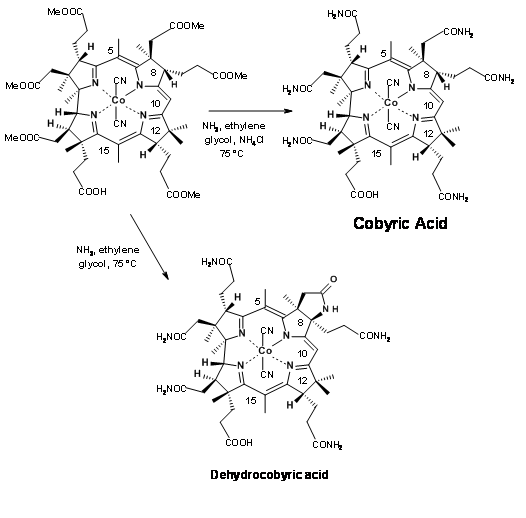
Finalmente se añadieron unos miligramos de acetato amónico a la mezcla de reacción para evitar la formación del anión amida, el cual se sospechó que era el culpable de la ciclación (Figura 13.22). Este truco ayudó. La reacción se completó dentro de las 10 horas, siendo el ácido cobirico el único producto con buenos rendimientos. Este ácido cobirico fue idéntico en todos los aspectos, particularmente en HPLC, con el producto natural, terminando así este largo viaje para la síntesis formal de la Vitamina B 12.
Esta extraordinaria aventura en la síntesis orgánica se destaca por varios logros más significativos.
- Desarrollos en la química de Corrin
- Estrategia sintética
- Desarrollo de varias metodologías nuevas
- Reglas de Woodward-Hoffmann
Incluso después de medio siglo esta síntesis de Vit B 12 sigue sin igual y sigue inspirando a generaciones de químicos.
Lectura adicional
- Avances recientes en la química de productos naturales, Woodward, R.B. Pure & Appl. Chem., 17, 519 (1968).
- Eschenmoser, A. Puro Appl. Chem., 297—316 (1963).
- Eschenmoser, A. Angew. Chem. Int. Ed., 5 (1988)
- Avances recientes en la química de productos naturales, Woodward, R.B. Pure & Appl. Chem., 25, 283 (1971).
- La síntesis total de vitamina B 1 2, R. B. Woodward, Pure & Appl. Chem. , 33, 145 (1973).
- Síntesis de Producto Natural y Vitamina B 1 2, Eschenmoser, A.; Wintner, C.E. Science, 196, 1410 (1977).
- Crowfoot-Hodgkin, D. et al. Naturaleza, 178, 64 (1956).
- Friedrich, W., Eds., Walter de Gruyter: Berlín, 37, (1979).
- Metas, Estrategias, Métodos. Weinheim, Alemania: VCH, (1996).
- El arte y la ciencia de la síntesis total en los albores del siglo XXI, K. C. Nicolaou, Dionisios Vourloumis, Nicolas Winssinger, y Phil S. Baran, Angew. Chem. Int. Ed., 39, 44 (2000).
- Vitamina B 12: Una aventura épica en síntesis total, de Neil Garg, 29 de enero de 2002.
- La síntesis total asimétrica de la vitamina B 1 2, Nathan S. Werner, Dinamarca Reunión del Grupo, 28 de septiembre de 2010.
- Nicolaou, K.C.; Sorensen, E. J. Vitamina B 1 2. Clásicos en Síntesis Total, VCH: Nueva York, 100, (2003).