
5.2: Isómeros configuracionales
- Page ID
- 73636

Iomerismo Geométrico
Hemos definido isómeros de manera muy general como moléculas no idénticas que poseen el mismo número y tipo de átomos. Sin embargo, hay varias formas en las que los isómeros pueden ser no idénticos. Entre los alquenos, el 1- y el 2-buteno son isómeros de posición, ya que en estos compuestos el doble enlace tiene una posición diferente en la cadena carbonada:
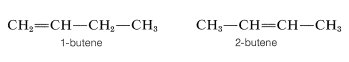
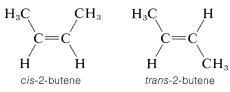
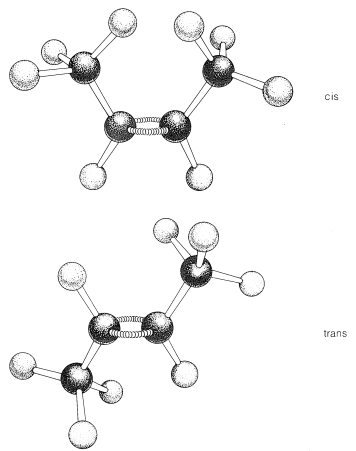
La mayoría, pero no todos los alquenos, tienen estereoisómeros que no son idénticos debido a las diferentes disposiciones espaciales de los átomos componentes. Así, existen dos estereoisómeros del 2-buteno que difieren en la disposición geométrica de los grupos unidos al doble enlace. En un isómero, ambos grupos metilo están en el mismo lado del doble enlace (cis-2-buteno) y en el otro, los grupos metilo están en lados opuestos del doble enlace (trans-2-buteno):
Debe quedar claro para usted que no habrá isómeros cis-trans de alquenos en los que un extremo del doble enlace lleve grupos idénticos. Por lo tanto, no esperamos que haya isómeros cis-trans de 1-buteno o 2-metilpropeno, y
de hecho ninguno se conoce:
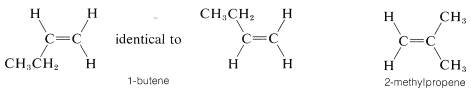
Es posible que desee verificar esto haciendo modelos de bolas y palos de estas sustancias.
La formación de anillos también confiere rigidez a la estructura molecular de tal manera que se evita la rotación alrededor de los enlaces del anillo. Como resultado, es posible el estereoisomería del tipo cis-trans. Por ejemplo, el 1,2-dimetilciclopropano existe en dos formas que difieren en la disposición de los dos grupos metilo con
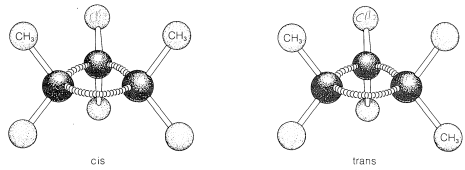
respeto al anillo. En el isómero cis, los grupos metilo se sitúan ambos por encima (o por debajo) del plano del anillo y en el isómero trans están situados uno arriba y uno abajo, como se muestra en la Figura 5-2. La interconversión de estos isómeros no ocurre sin romper uno o más enlaces químicos.
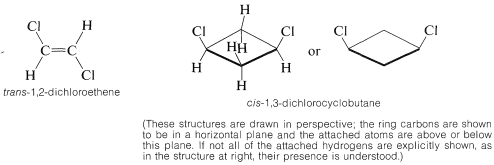
Los estereoisómeros que no se interconvierten rápidamente en condiciones normales, y por lo tanto son lo suficientemente estables como para separarse, específicamente se denominan isómeros configuracionales. Así, cis - y trans-2-buteno son isómeros configuracionales, al igual que cis - y trans-1,2-dimetilciclopropano. Los términos isomerismo cis-trans o isomería geométrica se utilizan comúnmente para describir isomería configuracional en compuestos con dobles enlaces y anillos. Al referirnos a la configuración de un isómero particular, nos referimos a especificar su geometría. Por ejemplo, el isómero de 1,2-dicloroeteno que se muestra a continuación tiene la configuración trans; el isómero de 1,3-diclorociclobutano tiene la configuración cis:
El isomerismo cis-trans se encuentra con mucha frecuencia. Por una convención, la configuración de un alqueno complejo se toma para corresponder a la configuración de la cadena continua más larga a medida que pasa a través del doble enlace. Así, el siguiente compuesto es trans-4-etil-3-metil-3-hepteno, a pesar de que dos grupos idénticos son cis uno con respecto al otro, debido a que la cadena continua más larga es trans a medida que pasa a través del doble enlace:
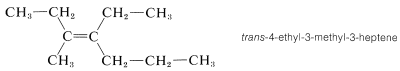
Observe que la isomería cis-trans no es posible en un triple enlace carbono-carbono, como para el 2-butino, porque la disposición de unión en los carbonos triples unidos es lineal:

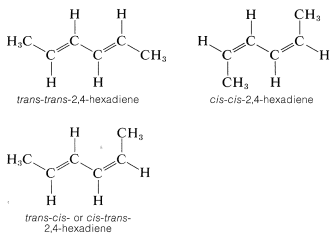
Muchos compuestos tienen más de un doble enlace y cada uno puede tener el potencial para la disposición cis o trans. Por ejemplo, el 2,4-hexadieno tiene tres configuraciones diferentes, las cuales se designan como trans-trans, cis-cis y trans-cis. Debido a que los dos extremos de esta molécula están sustituidos idénticamente, el trans-cis se vuelve idéntico al cis-trans:
Chiralidad
El tipo de estereoisomería más importante es el que surge cuando las moléculas poseen dos estructuras que no son idénticas y además son imágenes especulares entre sí. Este no es un concepto difícil ni desconocido. Muchas cosas que nos rodean, como nuestras manos, y pares de zapatos, no son idénticas y también son imágenes especulares unas de otras. De la misma manera, existen moléculas no idénticas en las que la única distinción entre ellas es que una es la imagen especular de la otra. Una afirmación común es que tales isómeros son imágenes especulares entre sí, pero estas imágenes no son “superponibles”. Un ejemplo sencillo de este tipo de estereoisomería es el 2-clorobutano , que puede existir en dos configuraciones espaciales,\(1\) y\(2\), que corresponden a reflexiones entre sí. Estos isómeros se denominan específicamente enantiómeros.
, que puede existir en dos configuraciones espaciales,\(1\) y\(2\), que corresponden a reflexiones entre sí. Estos isómeros se denominan específicamente enantiómeros.
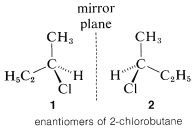
Se dice que los compuestos que carecen de simetría de reflexión -es decir, que no son idénticos a sus imágenes especulares- son quirales (pronunciado “ki-rall”, rimas con espiral). Este término se deriva de la palabra griega\(\chi \epsilon \iota \rho =\) mano; y “mandedness” o quiralidad es una propiedad de las moléculas asimétricas de tal manera que son posibles dos isómeros configuracionales que son imágenes especulares no idénticas. Se dice que los compuestos que poseen simetría de reflexión -es decir, que son idénticos a sus imágenes especulares- son aquirales. Los enantiómeros no son posibles para los compuestos aquirales. Un par enantiomérico es un par de sustancias cuyas moléculas son imágenes especulares no idénticas.
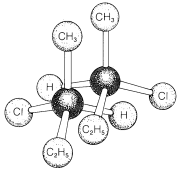
La cuestión apremiante en este punto es cómo podemos decir si una sustancia será quiral o aquiral. El origen más común de la quiralidad en las moléculas, y el que originalmente reconocieron van 't Hoff y Le Bel, es la presencia de uno o más átomos, generalmente átomos de carbono, cada uno de los cuales forma enlaces coplanares a cuatro átomos o grupos diferentes. Este es el caso del 2-clorobutano, porque el segundo carbono tetraédrico a lo largo de la cadena está unido a cuatro grupos diferentes: hidrógeno, cloro, metilo y etilo. Por lo tanto hay un par de enantiómeros,\(1\) y\(2\). Otro ejemplo es el 2-bromo-2-cloro-1,1,1-trifluoroetano, que es un anestésico de inhalación ampliamente utilizado. Los cuatro grupos diferentes en este caso son hidrógeno, cloro, bromo y trifluorometilo; el par de enantiómeros se muestra en Estructuras\(3\) y\(4\):

El átomo que lleva los cuatro sustituyentes diferentes en\(1\) y\(2\), o\(3\) y\(4\), se denomina átomos quirales o centro quiral. Este último es el término más general porque, como veremos más adelante (Sección 13-5A), la asimetría en las moléculas no necesita estar centrada en un átomo. \(^1\)
Al evaluar una estructura química para la quiralidad, debe buscar carbonos que lleven cuatro grupos unidos diferentes. Puede haber más de un carbono quiral, y debes estar alerta al hecho de que las diferencias estructurales en los grupos unidos no necesariamente aparecen en la primera, o incluso la segunda, posición a lo largo de una cadena. Como ejemplo, considere la quiralidad del 1,1,3-trimetilciclohexano,

Los carbonos\(C2\)\(C4\),\(C5\),, y\(C6\) son claramente aquirales porque cada uno está conectado a dos grupos idénticos, que para estos átomos son hidrógenos. Lo mismo es cierto\(C1\) porque está conectado a dos\(CH_3\) grupos. Se podría concluir que\(C3\) también es una posición aquiral porque está conectada a dos\(CH_2\) grupos. Pero esto estaría mal. Si miras más lejos, verás que los grupos adscritos a\(C3\) realmente son diferentes y son\(H\),\(CH_3\),\(-CH_2CH_2CH_2-\), y\(-CH_2C \left( CH_3 \right)_2\). Por lo tanto 1,1,3-trimetilciclohexano tiene un centro quiral en\(C3\). Por el contrario, el isómero 1,1,4 no tiene centros quirales porque los grupos unidos al anillo en\(C4\) son idénticos:
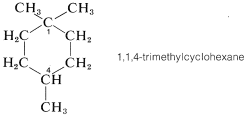
Otros términos que usaremos frecuentemente además de la quiralidad son mezcla racémica, resolución y racemización. Una mezcla de cantidades iguales de ambos enantiómeros es una mezcla racémica; la separación de una mezcla racémica en sus enantiómeros componentes es una resolución, y la conversión de cualquiera de los enantiómeros en partes iguales de ambos se llama racemización.
Actividad óptica
Hasta hace poco, el fenómeno de la quiralidad se conocía mejor como isomería óptica, y los isómeros configuracionales que son enantiómeros se denominaban antípodas ópticas. Las razones de esto son principalmente históricas. Se descubrió a principios del siglo XIX que muchos compuestos, ya sean sólidos, líquidos o gaseosos, tienen la propiedad de rotar el plano de polarización de la luz polarizada y se puede decir que son "ópticamente activos”. Una explicación satisfactoria del origen de la actividad óptica llegó mucho más tarde y se desarrolló en su forma moderna a partir de las investigaciones clásicas de Louis Pasteur, y del concepto de los átomos de carbono tridimensionales expresados independientemente por J. H. van 't Hoff y J. A. Le Bel. \(^2\)
La contribución de Pasteur a la estereoquímica vino como resultado de sus estudios sobre las formas de los cristales de ácido tartárico\(\ce{HO_2C-CHOH-CHOH-CO_2H}\), y sus sales. Se sabía que el ácido tartárico, subproducto de la producción de vino, era ópticamente activo, y Pasteur demostró que él, y diecinueve sales diferentes del mismo, formaban cristales que no eran idénticos a sus imágenes especulares. Se sabía que una sustancia diferente conocida como “ácido racémico”, para la que podemos escribir la misma fórmula condensada\(\ce{HO_2C-CHOH-CHOH-CO_2H}\), era ópticamente inactiva, y Pasteur esperaba que cuando cristalizara este ácido o sus sales obtendría cristales que serían idénticos a sus imágenes especulares. Sin embargo, la cristalización de la sal de sodio y amonio del ácido racémico a partir del agua a temperaturas inferiores\(28^\text{o}\) dio cristales de dos formas diferentes y estas formas fueron imágenes especulares entre sí. Pasteur recogió cuidadosamente los dos tipos de cristales y demostró que uno de ellos era idéntico a la sal correspondiente de ácido tartárico, excepto que giraba el plano de polarización de la luz polarizada en sentido contrario. Esta separación del ácido racémico en dos formas ópticamente activas ahora se llama una “resolución del ácido racémico”.
A partir de sus descubrimientos, Pasteur postuló que el “isomería óptica” tenía que relacionarse con la dismetría molecular de sustancias de tal manera que pudieran existir formas no idénticas de imagen especular. Sin embargo, quedó para van' t Hoff y Le Bel proporcionar, casi simultáneamente, una explicación satisfactoria a nivel molecular. En su primer trabajo publicado sobre carbono tetraédrico van 't Hoff dijo “... cada vez parece más que las presentes fórmulas constitucionales son incapaces de explicar ciertos casos de isomería; la razón de esto es quizás el hecho de que necesitamos una declaración más definida sobre las posiciones reales de los átomos”. \(^3\)Continúa discutiendo las consecuencias de las disposiciones tetraédricas de los átomos sobre el carbono, explícitamente en relación con el isomerismo óptico y el isomería geométrica, o cis-trans.
No es fácil para el químico de hoy apreciar plenamente las contribuciones de estos primeros químicos porque desde hace mucho tiempo aceptamos el carbono tetraédrico como un hecho establecido experimentalmente. En el momento en que se enunció el concepto, sin embargo, incluso la existencia de átomos y moléculas fue cuestionada abiertamente por muchos científicos, y atribuir “formas” a lo que en primer lugar parecían concepciones metafísicas era demasiado para que muchos aceptaran.
Propiedades de los Enantiómeros
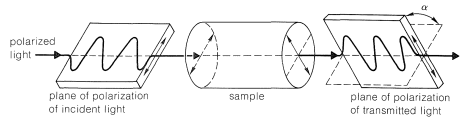
La actividad óptica es una propiedad experimentalmente útil y generalmente se mide como el ángulo de rotación (\(\alpha\)) del plano de polarización de la luz polarizada que pasa a través de soluciones de las sustancias bajo investigación (Figura 5-4). Donde la actividad óptica medible está presente, se encuentra que un enantiómero gira el plano de polarización en una dirección, mientras que la otra hace que el plano gire por igual pero en la dirección opuesta. Con referencia al plano de luz incidente, el enantiómero que gira el plano hacia la derecha se llama dextrorrotatorio y está simbolizado por d o (\(+\)); el enantiómero que gira el plano hacia la izquierda es levorrotatorio, simbolizado por l o ( \(-\)). Una mezcla racémica entonces puede ser designada como dl o (\(\pm\)), y no tendrá rotación óptica neta. Es muy importante saber que d, l, (\(+\)), o (\(-\)) no designan configuraciones. Por lo tanto, aunque (\(+\)) -2-butanol realmente tiene configuración\(5\) y (\(-\)) -2-butanol tiene configuración\(6\), no hay una manera simple de predecir que un signo particular de rotación se asociará con una configuración particular. Los métodos utilizados para asignar las configuraciones verdaderas a los enantiómeros se discutirán más adelante.
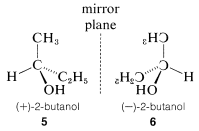
Un punto muy importante a tener en cuenta acerca de cualquier par de enantiómeros es que tendrán idénticas propiedades químicas y físicas, a excepción de los signos de sus rotaciones ópticas, con una condición importante: Todas las propiedades a comparar deben determinarse utilizando reactivos aquirales en un disolvente conformado de moléculas aquirales o, en definitiva, en un ambiente aquiral. Así, los puntos de fusión y ebullición (pero no las rotaciones ópticas) de\(5\) y\(6\) serán idénticos en un ambiente aquiral. Cómo un ambiente quiral o reactivos quirales influyen en las propiedades de sustancias como\(5\) y\(6\) serán consideradas en el Capítulo 19.
\(^1\)En la literatura más antigua, los centros quirales a menudo se denominan centros asimétricos y puede confundirse por la diferencia entre asimétricos y asimétricos. Tanto las moléculas asimétricas como las asimétricas (u objetos) son quirales. Un objeto asimétrico no tiene simetría en absoluto y se ve diferente desde todos los ángulos de visión. Fórmulas\(3\) y\(4\) representan moléculas asimétricas. Una molécula asimétrica es quiral, pero se ve igual desde más de un ángulo de visión. Un resorte helicoidal es asimétrico, se ve igual desde cada extremo. Nos encontraremos con moléculas asimétricas más adelante.
\(^2\)El carbono tetraédrico fue propuesto por primera vez por E. Paterno en 1869 (ver Sección1-1E), pero aparentemente no reconoció sus implicaciones para la quiralidad. Estas implicaciones fueron reconocidas primero por van 't Hoff y Le Bel, con van 't Hoff procediendo sobre la base de que los enlaces al carbono se dirigen a las esquinas de un tetraedro regular. Le Bel se opuso a una formulación tan rígida de los enlaces al carbono.
\(^3\)Un relato interesante y referencias a los primeros trabajos de van 't Hoff se pueden encontrar en “La recepción de J. H. van 't Hoff's Theory of the Asymmetric Carbon” de H. A. M. Snolder, J. Chem. Educ. 51, 2 (1974). Ha pasado un siglo desde que van 't Hoff publicó por primera vez su teoría, lo que hizo antes de obtener su doctorado en la Universidad de Utrecht. van 't Hoff fue el primer ganador del Premio Nobel de Química (1901) por su posterior trabajo en termodinámica y cinética química.
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."