
10.4: Adiciones electrofílicas a alquenos
- Page ID
- 73464

Las reacciones de los alcanos discutidas en el Capítulo 4 son procesos homolíticos, lo que significa que los enlaces se hacen y rompen a través de intermedios radicales o atómicos. Por el contrario, las\(E\) reacciones\(S_\text{N}\) y los haluros de alquilo, considerados en el Capítulo 8, implican escisión de enlaces heterolíticos y reactivos o productos iónicos. Un factor especialmente importante que contribuye a las diferencias entre las reacciones de los alcanos y haluros de alquilo es el ligero carácter iónico de los enlaces\(\ce{C-H}\) en comparación con los\(\ce{C}\) -haluro (ver Sección 1-3). Los alquenos son como los alcanos por ser compuestos no polares (Sección 4-1) y puede ser una sorpresa que muchas reacciones importantes de alquenos sean reacciones heterolíticas. ¿Por qué debería ser así? Sin duda porque los electrones en los dobles enlaces alquenos están más expuestos y accesibles que los electrones en un\(\ce{C-C}\) enlace alcano.
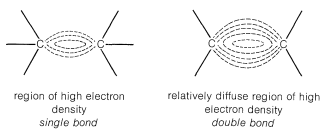
Esto es evidente a partir de los modelos atómico-orbitales de eteno descritos en la Sección 6-4C. Los electrones del doble enlace son empujados hacia afuera por sus repulsiones mutuas, y sus posiciones promedio están considerablemente más lejos del eje del enlace que las posiciones electrónicas de un enlace simple (Figura 10-6). En tales circunstancias, se espera que los reactivos electrofílicos, que actúan para adquirir electrones en reacciones químicas (Sección 8-1), sean particularmente reactivos. Este es en realidad el caso. Además, los reactivos que son principalmente nucleofílicos (donadores de electrones) son notoriamente pobres para iniciar reacciones en dobles enlaces carbono-carbono. Las excepciones ocurren cuando los dobles enlaces llevan sustituyentes con un grado suficientemente alto de potencia de atracción de electrones para reducir la densidad de electrones en el doble enlace lo suficiente como para permitir el ataque de un agente nucleófilo.
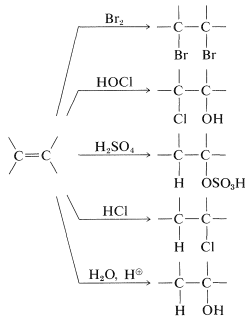
Ejemplos de reactivos electrofílicos que normalmente se añaden a dobles enlaces carbono-carbono de alquenos para dar compuestos saturados incluyen halógenos (\(\ce{Cl_2}\)\(\ce{Br_2}\), y\(\ce{I_2}\)), haluros de hidrógeno (\(\ce{HCl}\)y\(\ce{HBr}\)), ácidos hipohalosos (\(\ce{HOCl}\)y\(\ce{HOBr}\)), agua y ácido sulfúrico:
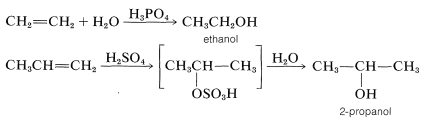
Los mecanismos de estas reacciones tienen mucho en común y han sido estudiados extensamente desde este punto de vista. También tienen una utilidad sintética muy considerable. La adición de agua a los alquenos (hidratación) es particularmente importante para la preparación de una serie de alcoholes comercialmente importantes. Así, el etanol y el 2-propanol (alcohol isopropílico) se elaboran a gran escala mediante la hidratación de los alquenos correspondientes (eteno y propeno) utilizando como catalizadores ácidos sulfúrico o fosfórico. La naturaleza de este tipo de reacción se describirá más adelante.
El mecanismo iónico paso a paso, adición de halógeno
Daremos particular atención aquí a la adición de bromo a los alquenos porque esta reacción se lleva a cabo muy convenientemente en el laboratorio e ilustra una serie de puntos importantes sobre las reacciones de adición electrófila. Gran parte de lo que sigue se aplica a la adición de los otros halógenos, excepto el flúor.
Una observación significativa con respecto a la adición de bromo es que ésta y muchas de las otras reacciones enumeradas anteriormente proceden en la oscuridad y no están influenciadas por inhibidores radicales. Esto es evidencia contra un mecanismo de cadena radical del tipo involucrado en la halogenación de alcanos (Sección 4-4D). Sin embargo, no excluye la operación de reacciones de adición radical bajo otras condiciones y, como veremos más adelante en este capítulo, bromo, cloro y muchos otros reactivos que comúnmente se agregan a los alquenos por mecanismos iónicos también pueden agregar por mecanismos radicales.
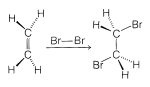
Una alternativa a una reacción radical-cadena para la adición de bromo a un alqueno sería el proceso simple de cuatro centros y una etapa que se muestra en la Figura 10-7.
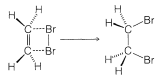
El mecanismo de la Figura 10-7 no puede ser correcto para la adición de bromo a los alquenos en solución por dos razones importantes. Primero, observe que este mecanismo requiere que los dos\(\ce{C-Br}\) enlaces se formen en el mismo lado del doble enlace, y por lo tanto produzcan adición suprafacial. Sin embargo, hay mucha evidencia que demuestra que el bromo y muchos otros reactivos se suman a los alquenos para formar productos de adición antarafaciales (Figura 10-8).
El ciclohexeno añade bromo para dar trans-1,2-dibromociclohexano:
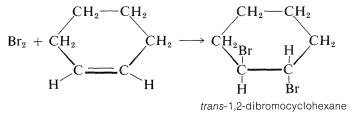
El isómero cis no se forma en absoluto. Para dar el isómero trans, los dos nuevos\(\ce{C-Br}\) enlaces tienen que formarse en lados opuestos del doble enlace por adición antarafacial. Pero esto es imposible por un mecanismo de un solo paso porque el\(\ce{Br-Br}\) enlace tendría que estirarse demasiado para permitir la formación de ambos\(\ce{C-Br}\) enlaces al mismo tiempo.
La segunda evidencia contra el mecanismo de la Figura 10-7 es que las reacciones de adición de bromo llevadas a cabo en presencia de más de un reactivo nucleófilo suelen dar mezclas de productos. Así, la adición de bromo a una solución de alqueno en metanol que contiene cloruro de litio conduce no solo al dibromoalcano esperado, sino también a productos resultantes del ataque por iones cloruro y por el disolvente:
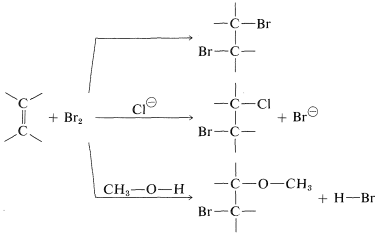
La intervención de nucleófilos extraños sugiere un mecanismo paso a paso en el que los nucleófilos compiten por un intermedio reactivo formado en una de las etapas.
Para la adición de bromo al eteno se ilustra un mecanismo de dos pasos algo sobre-simplificado que da cuenta de la mayoría de los hechos anteriores. [En la formación que se muestra a continuación, no se considera que las flechas curvas tengan un significado mecanicista real, sino que se utilizan principalmente para mostrar qué átomos pueden considerarse nucleófilos (donar electrones) y cuáles como electrófilos (aceptar electrones). Las puntas de flecha siempre deben ser dibujadas para señalar los átomos que se formulan como aceptando un par de electrones.]
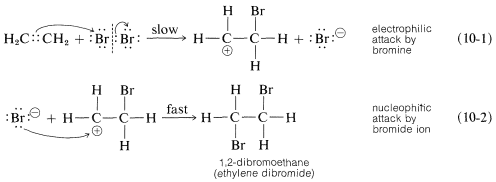
El primer paso (que implica el ataque electrófilo por bromo en el doble enlace) produce un ion bromuro y un carbocatión, como se muestra en la Ecuación 10-1. \(^1\)
Como sabemos por nuestro estudio de\(S_\text{N}1\) reacciones (Sección 8-4), los carbocationes reaccionan fácilmente con reactivos nucleofílicos. Por lo tanto, en la segunda etapa del mecanismo de adición de bromo, mostrado en la Ecuación 10-2, se espera que el catión bromoetilo se combine rápidamente con el ion bromuro para dar el compuesto dibromo. Sin embargo, si otros nucleófilos, como\(\ce{Cl}^\ominus\) o\(\ce{CH_3OH}\), están presentes en solución, deberían poder competir con el ion bromuro por el catión, como en las Ecuaciones 10-3 y 10-4, y las mezclas de productos resultarán:
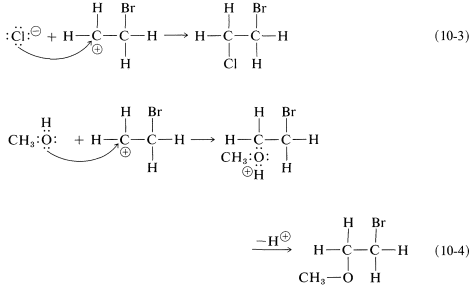
Para dar cuenta de la observación de que todas estas reacciones resultan en la adición antarafacial, debemos concluir que los primeros y segundos pasos tienen lugar desde lados opuestos del doble enlace.
¿Por qué Adición Antarafacial?
El intermedio de carbocatión simple de la Ecuación 10-1 no tiene en cuenta la formación del producto de adición antarafacial. Los resultados con\(S_\text{N}1\) reacciones (Sección 8-6) y la representación atómica-orbital (ver Sección 6-4E) predicen que los enlaces al átomo de carbono cargado positivamente de un carbocatión deben estar en un plano. Por lo tanto, en la segunda etapa de adición de bromo a cicloalquenos, el ion bromuro podría atacar cualquier lado del carbono positivo plano para dar una mezcla de cis - y trans -1,2-dibromociclohexanos. Sin embargo, la adición antarafacial ocurre exclusivamente:
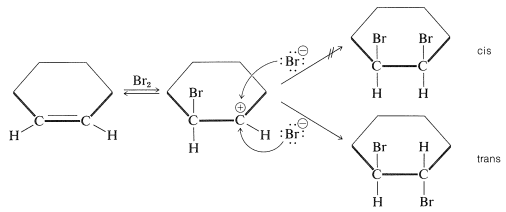
Para dar cuenta de la estereoespecificidad de la adición de bromo a los alquenos, se ha sugerido que en el ataque electrófilo inicial del bromo se forma un intermedio cíclico que tiene bromo unido a ambos carbonos del doble enlace. Tal ion “puenteado” se llama ion bromo porque el bromo lleva formalmente la carga positiva:
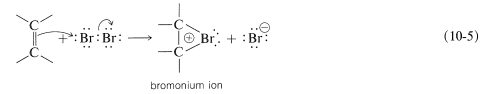
Un\(S_\text{N}2\) tipo de ataque de ion bromuro, u otro nucleófilo, en el carbono en el lado opuesto al grupo puente entonces da como resultado la formación del producto de adición antarafacial:
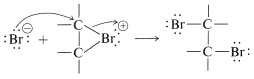
Puede parecer que nos hemos contradido porque la Ecuación 10-1 muestra un carbocatión que se forma en la adición de bromo, pero la Ecuación 10-5 sugiere un ion bromo. En realidad, la formulación de intermedios en reacciones de adición de alquenos como iones “abiertos” o como iones cíclicos es un tema controvertido, incluso después de muchos años de estudio. Desafortunadamente, no es posible determinar la estructura de los iones intermedios por ningún método físico directo porque, bajo las condiciones de la reacción, los iones son tan reactivos que forman productos más rápidamente de lo que pueden observarse. Sin embargo, es posible generar iones bromonio estables, así como los correspondientes iones cloronio y yodonio. La técnica consiste en utilizar bajas temperaturas en ausencia de nucleófilos fuertes y comenzar con un 1,2-dihaloalcano y pentafluoruro de antimonio en dióxido de azufre líquido:
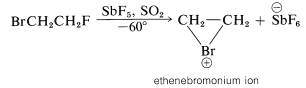
Los\(\ce{C_2H_4Br}^\oplus\) iones producidos de esta manera son relativamente estables y se ha demostrado por RMN que tienen la estructura de iones halonio cíclicos.
Complejos de agentes electrofílicos con dobles enlaces
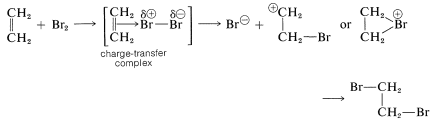
Hay un aspecto adicional de las adiciones polares a los alquenos que debemos considerar, a saber, que los reactivos electrófilos forman complejos sueltos con los\(\pi\) electrones de los dobles enlaces de los alquenos antes de la reacción por adición. Los complejos de este tipo se denominan complejos de transferencia de carga (o \(\pi\)complejos). La formación de un complejo entre yodo y ciclohexeno se demuestra por el hecho de que el yodo se disuelve en ciclohexeno para dar una solución marrón, mientras que sus soluciones en ciclohexano son violetas. La solución marrón de yodo en ciclohexeno se desvanece lentamente a medida que se produce la adición para dar trans-1,2-diyodociclohexano incoloro.
Las estructuras precisas de Lewis no se pueden escribir para complejos de transferencia de carga, pero comúnmente se representan como
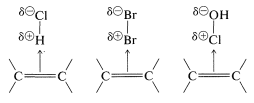
con la flecha que denota que los electrones del doble enlace están asociados con el electrófilo. Estos complejos representan probablemente la primera etapa en la formación de productos de adición mediante una secuencia tal como la siguiente para la adición de bromo:
Adición de ácidos protónicos
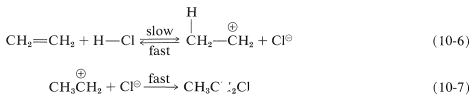
Hemos visto que los electrófilos pueden reaccionar con alquenos para formar enlaces carbono-halógeno donando halógeno positivo,\(\ce{Br}^\oplus\),\(\ce{Cl}^\oplus\), o\(\ce{I}^\oplus\). Asimismo, los enlaces carbono-hidrógeno pueden estar formados por donantes de protones apropiadamente fuertes, que, por supuesto, son típicamente ácidos protónicos fuertes. Estos ácidos son más efectivos en ausencia de grandes cantidades de agua debido a que el agua puede competir con el alqueno como aceptor de protones (ver también Sección 10-3E). La adición de cloruro de hidrógeno al eteno se produce por medio de una etapa de transferencia de protones para dar el catión etilo y un ion cloruro (Ecuación 10-6) seguido de una etapa en la que el ion cloruro nucleófilo se combina con el catión etilo (Ecuación 10-7):
Todos los haluros de hidrógeno\(\ce{HF}\),,\(\ce{HCl}\)\(\ce{HBr}\), y\(\ce{HI}\)) se agregarán a los alquenos. La adición de fluoruro de hidrógeno, aunque es fácil, es fácilmente reversible. Sin embargo, una solución de fluoruro de hidrógeno\(70\%\) anhidro y\(30\%\) de la base orgánica débil, piridina, que es aproximadamente 1/10,000 veces más fuerte que el amoníaco, funciona mejor, y con ciclohexeno da fluorociclohexano. Con el yoduro de hidrógeno, se debe tener cuidado para evitar los productos de\(\ce{I_2}\) adición resultantes del yodo formado por reacciones de oxidación como
\[4 \ce{HI} + \ce{O_2} \rightarrow 2 \ce{I_2} + 2 \ce{H_2O}\]
Con bromuro de hidrógeno, la adición de cadena radical puede intervenir a menos que las condiciones de reacción se controlen cuidadosamente (esto se discutirá en la Sección 10-7).
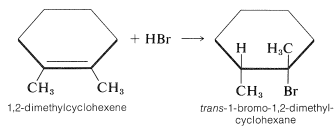
La estereoquímica de la adición depende en gran medida de la estructura del alqueno, pero para los alquenos simples y cicloalquenos, la adición ocurre predominantemente de manera antarafacial. Por ejemplo, el bromuro de hidrógeno reacciona con 1,2-dimetilciclohexeno para dar el producto de adición antarafacial:
Hidratación
Anteriormente mencionamos que la hidratación de alquenos requirió un ácido fuerte como catalizador, ya que el agua misma es un ácido demasiado débil para iniciar la etapa de transferencia de protones. Sin embargo, si está presente una pequeña cantidad de un ácido fuerte como el ácido sulfúrico, se forman iones hidronio\(\ce{H_3O}^\oplus\), en cantidad suficiente para protonar alquenos razonablemente reactivos, aunque de ninguna manera tan efectiva como lo hace el ácido sulfúrico concentrado. El carbocatión formado entonces es atacado rápidamente por una molécula nucleofílica de agua para dar el alcohol como su ácido conjugado, el\(^2\) cual regenera el ion hidronio transfiriendo un protón al agua. La secuencia de reacción sigue para 2-metilpropeno:
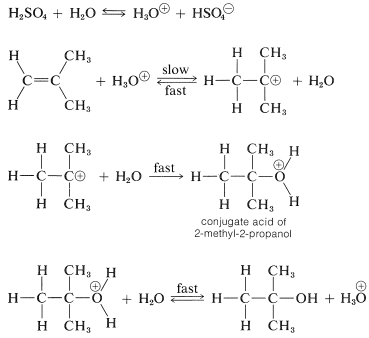
En esta secuencia, el ácido actúa como catalizador debido a que el ion hidronio usado en la etapa de adición de protones se regenera en la etapa final.
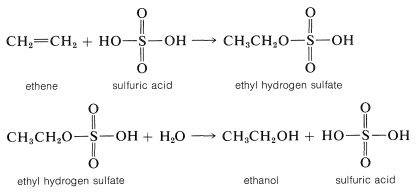
Se prefiere el ácido sulfúrico (o ácido fosfórico) como catalizador ácido para la adición de agua a los alquenos debido a que la base conjugada,\(\ce{HSO_4-}\) (o\(\ce{H_2PO_4-}\)), es un nucleófilo pobre y no interfiere en la reacción. Sin embargo, si la concentración de agua se mantiene baja mediante el uso de ácido concentrado, se produce la adición para dar ésteres de sulfato (o fosfato). Los ésteres formados con ácido sulfúrico son sulfatos de ácido alquílico\(\ce{R-OSO_3H}\) o sulfatos de dialquilo\(\ce{(RO)_2SO_2}\). De hecho, esta es una de las principales vías utilizadas en la producción comercial de etanol y 2-propanol. Ethen y ácido sulfúrico dan hidrógeno sulfato de etilo, que reacciona fácilmente con agua en una segunda etapa para dar etanol:
Ácidos Acuosos versus No Acuosos. Fortalezas a los ácidos
Una de las características más confusas de la química orgánica es la multitud de condiciones que se utilizan para llevar a cabo un determinado tipo de reacción, como la adición electrófila de ácidos protónicos a diferentes alquenos. Ácidos fuertes, ácidos débiles, agua, sin agua - ¿Por qué no puede haber un procedimiento estándar? El problema es que los alquenos tienen tendencias muy diferentes para aceptar protones. En la fase vapor,\(\Delta H^0\) para la adición de un protón al eteno es aproximadamente\(35 \: \text{kcal}\) más positiva que para el 2-metilpropeno, y aunque la diferencia debería ser menor en solución, todavía sería grande. Por lo tanto, podemos anticipar (y encontramos) que se necesita un donante de protones mucho más poderoso para iniciar la adición de un ácido al eteno que al 2-metilpropeno. Pero, ¿por qué no usar en todos los casos un ácido lo suficientemente fuerte como para protonar cualquier alqueno al que se quiera agregar un ácido protónico? Dos razones: Primero, los ácidos fuertes pueden inducir reacciones secundarias indeseables, por lo que uno generalmente intentará no usar un ácido más fuerte de lo necesario; segundo, ¡un ácido muy fuerte puede incluso evitar que ocurra la reacción deseada!
En química elemental, generalmente tratamos con ácidos en solución acuosa más o menos diluida y pensamos que los ácidos sulfúrico, clorhídrico y nítrico son igualmente fuertes porque cada uno está esencialmente completamente disociado en solución de agua diluida:
\[\ce{HCl} + \ce{H_2O} \overset{\longrightarrow}{\leftarrow} \ce{H_3O}^\oplus + \ce{Cl}^\ominus\]
Esto no quiere decir que en realidad sean ácidos igualmente fuertes. Significa sólo que cada uno de los ácidos es lo suficientemente fuerte como para donar todos sus protones al agua. Podemos decir que el agua tiene un “efecto nivelador” sobre las fuerzas ácidas porque mientras un ácido pueda donar sus protones al agua, la solución solo tiene una “fuerza” ácida que está determinada por la\(\ce{H_3O}^\oplus\) concentración, porque\(\ce{H_3O}^\oplus\) es donde están los protones.
Ahora bien, si usamos aceptores de protones más pobres como solventes, encontramos que los poderes donadores de protones de varios ácidos “fuertes” comienzan a extenderse inmensamente. Además, empiezan a suceder cosas nuevas. Por ejemplo, el eteno no se hidrata apreciablemente por un ácido acuoso diluido; simplemente es demasiado difícil transferir un protón del ion hidronio al eteno. Por lo que usamos ácido sulfúrico concentrado, que es lo suficientemente fuerte como para agregar un protón al eteno. Pero ahora no conseguimos hidratación, porque cualquier agua que esté presente en el ácido sulfúrico concentrado es prácticamente toda convertida en\(\ce{H_3O}^\oplus\), ¡lo cual no es nucleofílico!
\[\ce{H_2SO_4} + \ce{H_2O} \rightarrow \ce{H_3O}^\oplus + \ce{HSO_4-}\]
Sin embargo, la formación de\(\ce{H_3O}^\oplus\) conduce a la formación de\(\ce{HSO_4-}\), que tiene suficiente carácter nucleofílico para reaccionar con el\(\ce{CH_3CH_2+}\) para dar hidrógeno sulfato de etilo y éste se forma en lugar del ácido conjugado de etanol (Sección 10-3E). El epítome del uso de ácido más fuerte y nucleófilo más débil es con líquido\(\ce{SO_2}\) (pb\(\sim 10^\text{o}\)) como disolvente y\(\ce{HBF_6}\) como ácido. Este disolvente es un aceptor de protones muy pobre (lo que significa que su ácido conjugado es un donador de protones muy bueno) y\(\ce{SbF_6-}\) es un nucleófilo extremadamente pobre. Si añadimos eteno a tal solución,\(\ce{CH_3CH_2+} \ce{SbF_6-}\) se forma una solución estable de. La razón es que no hay mejor aceptor de protones presente que\(\ce{CH_2=CH_2}\) y ningún nucleófilo lo suficientemente bueno como para combinarse con el catión.
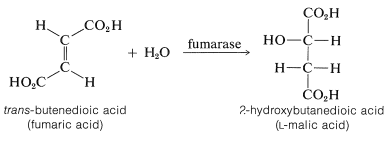
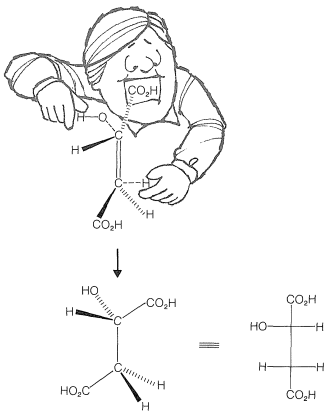
Una reacción de hidratación biológica
La conversión de ácido fumárico en ácido málico es una reacción de hidratación biológica importante. Es uno de un ciclo de reacciones (ciclo del ácido cítrico de Krebs) involucradas en la combustión metabólica de combustibles (aminoácidos e hidratos de carbono) hacia\(\ce{CO_2}\) y\(\ce{H_2O}\) en una célula viva.
\(^1\)Una alternativa a la Ecuación 10-1 sería haber\(\ce{Br_2}\) ionizado\(\ce{Br}^\oplus\) y\(\ce{Br}^\ominus\), con un ataque posterior de\(\ce{Br}^\oplus\) sobre el doble enlace para producir el carbocatión. El hecho es que la energía requerida para tal ionización de\(\ce{Br_2}\) es prohibitivamente grande incluso en solución de agua\(\left( \Delta H^0 \geq 80 \: \text{kcal} \right)\). Uno podría preguntarse por qué la Ecuación 10-1 podría ser más favorable. El calculado\(\Delta H^0\) para\(\ce{CH_2=CH_2} + \ce{Br_2} \rightarrow \cdot \ce{CH_2-CH_2Br} + \ce{Br} \cdot\) es\(+41 \: \text{kcal}\), que sólo es ligeramente más favorable que el\(\Delta H^0\) para\(\ce{Br_2} \rightarrow 2 \ce{Br} \cdot\) de\(46.4 \: \text{kcal}\). Sin embargo, los datos termoquímicos disponibles sugieren que la facilidad de transferir un electrón de\(\cdot \ce{CH_2CH_2Br}\)\(\ce{Br} \cdot\) a para dar\(^\oplus \ce{CH_2CH_2Br} + \ce{Br}^\ominus\) es aproximadamente\(80 \: \text{kcal}\) más favorable que\(2 \ce{Br} \cdot \rightarrow \ce{Br}^\oplus + \ce{Br}^\ominus\). Por lo tanto, es probable que el conjunto\(\Delta H^0\) de la Ecuación 10-1 sea aproximadamente\(85 \: \text{kcal}\) más favorable que\(\ce{Br_2} \rightarrow \ce{Br}^\oplus + \ce{Br}^\ominus\).
\(^2\)Los términos ácido conjugado y base conjugada son muy convenientes para designar sustancias que son difíciles de nombrar simplemente como ácidos, bases o sales. El ácido conjugado de un compuesto\(\ce{X}\) es\(\ce{XH}^\oplus\) y la base conjugada de\(\ce{HY}\) es\(\ce{Y}^\ominus\). Así\(\ce{H_3O}^\oplus\) es el ácido conjugado del agua, mientras que\(\ce{OH}^\ominus\) es su base conjugada. El agua en sí es entonces tanto la base conjugada\(\ce{H_3O}^\oplus\) de como el ácido conjugado de\(\ce{OH}^\ominus\)
Referencias
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."