
18.9: Reacciones en el\(\alpha\) Carbons of Carboxylic Acid Derivatives
- Page ID
- 72906

Las propiedades ácidas de los ésteres con\(\alpha\) hidrógenos
Muchas reacciones sintéticas importantes en las que se forman\(\ce{C-C}\) enlaces involucran ésteres y son provocadas por reactivos básicos. Esto es posible porque los\(\alpha\) hidrógenos de un éster, tal como\(\ce{RCH_2CO_2C_2H_5}\), son débilmente ácidos, y una base fuerte, como el etóxido de sodio, puede producir una concentración significativa del anión éster en equilibrio:
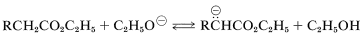
La acidez de\(\alpha\) los hidrógenos se atribuye en parte a los efectos inductivos de atracción de electrones de los oxígenos del éster, y en parte a la estabilización por resonancia del anión resultante (Sección 17-1A):
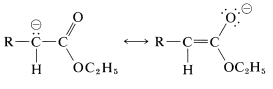
Cuando el\(\alpha\) carbono del éster lleva un segundo grupo fuertemente atrayente de electrones, la acidez del\(\alpha\) hidrógeno aumenta enormemente. Ejemplos de tales compuestos siguen:
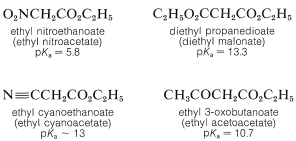
La estabilización de los aniones de estos ésteres especialmente activados es mayor que para los ésteres simples debido a los efectos inductivos de extracción de electrones de los sustituyentes, pero lo que es más importante porque la carga negativa puede distribuirse en más de dos centros. Así, para el anión del 3-oxobutanoato de etilo podemos considerar las tres estructuras de enlace de valencia,\(14a\) a través de\(14c\), como importantes para contribuir al híbrido,\(14\):
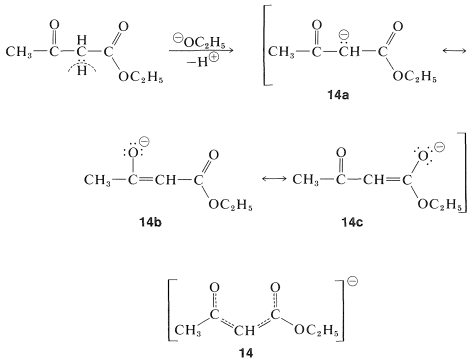
El anión\(14\),, es suficientemente estable en relación con el éster que\(K_\text{a}\) está aproximadamente\(10^{-11}\) en solución acuosa (Cuadro 17-1).
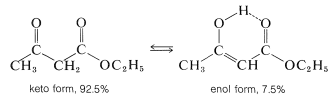
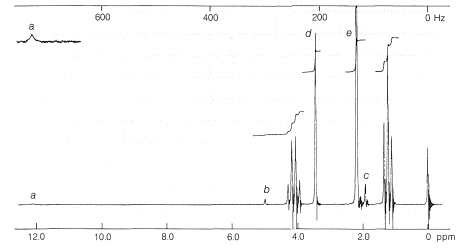
El 3-oxobutanoato de etilo existe a temperatura ambiente como una mezcla de equilibrio de tautómeros cetogénicos y enoles en la proporción de 92.5 a 7.5. La presencia de enol se puede mostrar por titulación rápida con bromo, pero es más evidente a partir del espectro de RMN protónica (Figura 18-6), que muestra la absorción de los protones hidroxilo, alquenil y metilo de la forma enol, además de las absorciones esperadas para la forma keto:
La interconversión de las formas enol y cetodel 3-oxobutanoato de etilo es catalizada poderosamente por las bases a través del anión\(15\), y menos por los ácidos a través del ácido conjugado de la forma ceto-forma:
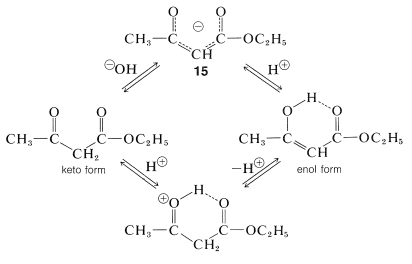
Sin embargo, si el contacto con sustancias ácidas y básicas se excluye rígidamente hasta el punto de usar equipos de cuarzo en lugar de vidrio (el vidrio normalmente tiene una superficie ligeramente alcalina), entonces la interconversión es lo suficientemente lenta como para que sea posible separar el enol de menor punto de ebullición de la forma cetofrcional destilación a presión reducida. Los isómeros separados son indefinidamente estables cuando se almacenan\(-80^\text{o}\) en recipientes de cuarzo.
La condensación de Claisen
Una de las reacciones más útiles de los ésteres inducidas por bases se ilustra por la autocondensación del etanoato de etilo bajo la influencia del etóxido de sodio para dar 3-oxobutanoato de etilo:
\[\ce{CH_3CO_2C_2H_5} + \ce{H-CH_2CO_2C_2H_5} \overset{^\ominus \ce{OC_2H_5}}{\longrightarrow} \ce{CH_3COCH_2CO_2C_2H_5} + \ce{C_2H_5OH}\]
Esta reacción, llamada la condensación de Claisen, es interesante porque, a partir de la consideración de las energías de enlace y estabilización, se espera que sea desfavorable termodinámicamente con\(\Delta H^0\) (vapor) igual a\(6 \: \text{kcal mol}^{-1}\). Esta expectativa se realiza en la práctica, y se ha invertido mucho esfuerzo para determinar las condiciones mediante las cuales se pueden obtener rendimientos prácticos del producto de condensación.
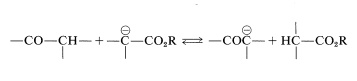
La condensación de Claisen se asemeja tanto a la adición de aldol (Sección 17-3) como a las adiciones de carbonilo de derivados ácidos discutidos previamente (Secciones 16-4 y 18-7). El primer paso, como se muestra en la Ecuación 18-10, es la formación del anión de etanoato de etilo que, al ser un potente nucleófilo, ataca el carbono carbonilo de una segunda molécula de éster (Ecuación 18-11). La eliminación del ion etóxido conduce entonces al\(\beta\) -ceto-éster, 3-oxobutanoato de etilo (Ecuación 18-12):
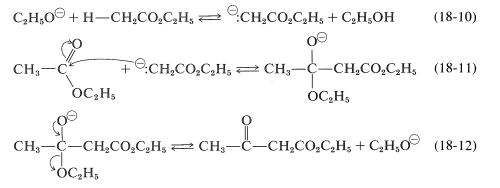
La suma de estos pasos representa un equilibrio desfavorable, y solo se obtienen rendimientos satisfactorios del\(\beta\) -ceto-éster si el equilibrio puede ser desplazado por la eliminación de uno de los productos.
Una forma sencilla de hacer esto es eliminar el etanol por destilación a medida que se forma; sin embargo, esto puede ser difícil de llevar hasta su finalización y, en cualquier caso, es contraproducente si el éster de partida es de bajo punto de ebullición. Alternativamente, se puede usar un gran exceso de etóxido de sodio. Esto es útil porque el etanol es un ácido más débil que el éster enol, y el exceso de etóxido desplaza el equilibrio hacia la derecha a través de la conversión del\(\beta\) -ceto-éster en la sal de enolato (Ecuación 18-13).
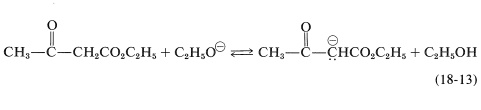
Obviamente, el producto de condensación debe recuperarse de la sal de enol y aislarse en condiciones que eviten la reversión a los materiales de partida. El mejor procedimiento es apagar la mezcla de reacción vertiéndola en un exceso de ácido diluido en frío.
Una limitación en la condensación de Claisen es que aunque el éster de partida necesita tener solo un\(\alpha\) hidrógeno para que ocurran las Reacciones 18-10 a 18-12, son necesarios dos\(\alpha\) hidrógenos para un equilibrio favorable utilizando la reacción de ionización de la Ecuación 18-13. Como resultado, no es sorprendente encontrar que el 2-metilpropanoato de etilo no se condensa consigo mismo en presencia de etóxido de sodio, debido a que el producto de condensación no tiene\(\alpha\) hidrógeno junto al grupo éster:
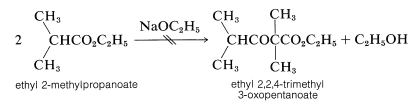
Sin embargo, se usa un exceso de una base mucho más fuerte que el etóxido de sodio [como trifenilmetilsodio,\(\ce{(C_6H_5)_3C}^\ominus \ce{Na}^\oplus\)], esta misma condensación sí se produce en rendimientos razonables. La razón es que la base ahora es lo suficientemente fuerte como para convertir el alcohol formado en la reacción en etóxido de sodio, desplazando así el equilibrio hacia la derecha:
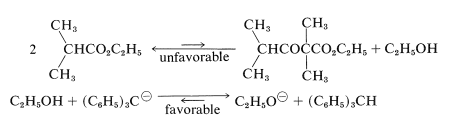
La reacción general entonces es
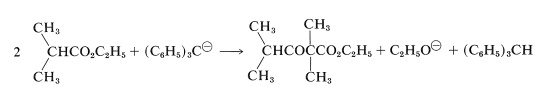
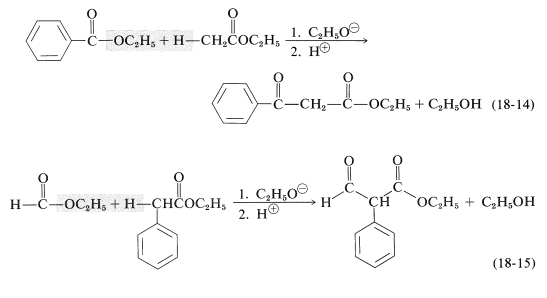
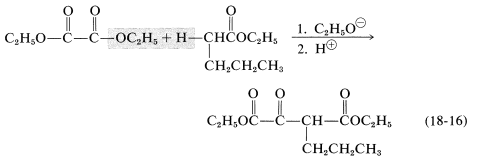
Las condensaciones de claisen se pueden llevar a cabo entre dos ésteres diferentes pero, debido a que hay cuatro productos posibles, a menudo resultan mezclas. Se encuentra menos dificultad si uno de los ésteres no tiene\(\alpha\) hidrógeno y reacciona fácilmente con un carbanión de acuerdo con las Ecuaciones 18-11 y 18-12. La reacción tiene entonces un parecido considerable con las adiciones mixtas de aldol discutidas en la Sección 17-3C. Entre los ésteres útiles sin\(\alpha\) hidrógenos, y con la reactividad electrófila requerida, se encuentran los de los ácidos bencenocarboxílico, metanoico, etanodioico y carbónico. Varios ejemplos prácticos de condensaciones mixtas de Claisen se muestran en las Ecuaciones 18-14 a 18-16 (todos los productos existen en la medida de\(10\%\) las formas enol):
Una variación importante en la condensación de Claisen es usar una cetona como reactivo aniónico. Esto suele funcionar bien porque las cetonas suelen ser más ácidas que los ésteres simples y la autocondensación inducida por bases de cetonas (adición aldólica) es termodinámicamente desfavorable (Sección 17-3C). Un ejemplo típico es la condensación de ciclohexanona con etanodioato de dietilo (oxalato de dietilo):

\(\alpha\)-Los ésteres cetogénicos del tipo formado según las Ecuaciones 18-16 y 18-17 tienen utilidad sintética ya que pierden monóxido de carbono cuando se calientan fuertemente:
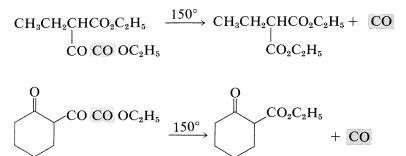
Anteriormente se mencionó una reacción de descarbonilación algo similar para la difenilpropanotriona (Sección 17-10).
Alquilación de Aniones Ester
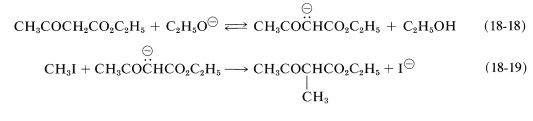
Los aniones de ésteres como 3-oxobutanoato de etilo y propanodioato de dietilo pueden alquilarse con haluros de alquilo. Estas reacciones son importantes para la síntesis de ácidos carboxílicos y cetonas y son de carácter similar a la alquilación de cetonas discutida anteriormente (Sección 17-4A). El éster es convertido por una base fuerte en el anión enolato, Ecuación 18-18, que luego es alquilado en una\(S_\text{N}2\) reacción con el haluro de alquilo, Ecuación 18-19. Por lo general, predomina\(\ce{C}\) la -alquilación:
Los ésteres del ácido propanodioico (malónico) pueden alquilarse de manera similar (Ecuación 18-20):
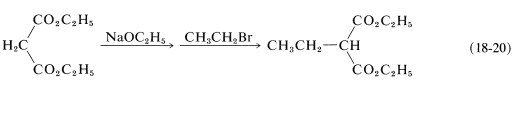
Desafortunadamente, la monoalquilación rara vez ocurre limpiamente por la secuencia anterior siempre que el producto de monoalquilación tenga un\(\alpha\) hidrógeno localizado para permitir que se produzca la dialquilación. En la práctica, las reacciones de alquilación, usando un mol de éster, un mol de etóxido de sodio y un mol de un haluro de alquilo (por ejemplo,\(\ce{CH_3I}\)), dan una mezcla del éster de partida, sus productos de mono- y dialquilación. La situación es más favorable cuando se introducen grandes grupos alquilo, porque entonces las propiedades físicas y reactividades de los materiales de partida y de los productos de mono- y dialquilación difieren considerablemente. Por lo general, la dialquilación se inhibe al tener un grupo alquilo voluminoso en el producto de monoalquilación.
Los ésteres 3-oxobutanoicos y propanodioicos sustituidos con alquilo pueden hidrolizarse bajo condiciones ácidas a los ácidos correspondientes, y cuando estos se calientan se descarboxilan fácilmente (Sección 18-4). Los ésteres alquílicos 3-oxobutanoicos producen así metil alquil cetonas, mientras que los ésteres alquilpropanodioicos producen ácidos carboxílicos:
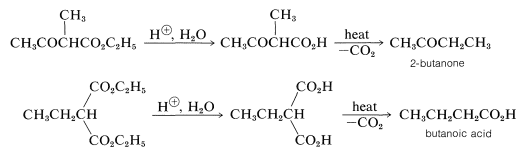
Estas reacciones se conocen comúnmente como síntesis de acetoacético-éster cetona y ácido malónico-éster, respectivamente.
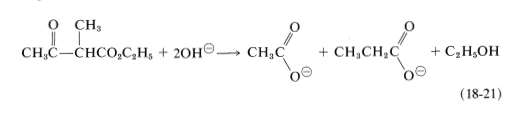
Los ésteres alquílicos 3-oxobutanoicos reaccionan con álcali concentrado por un camino diferente para revertir la condensación de Claisen:
Acilación de Aniones Ester
Los aniones enolados de ésteres, tales como 3-oxobutanoato de etilo o propanodioato de dietilo, reaccionan con haluros o anhidridos de acilo para dar productos de acilación. Estas reacciones se llevan a cabo mejor usando hidruro de sodio en lugar de etóxido de sodio para la producción de la sal de enol, porque entonces no se libera alcohol para reaccionar con el haluro o anhídrido de acilo:
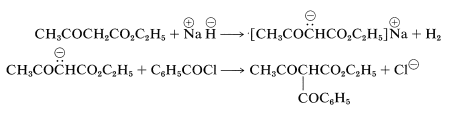
Adiciones de tipo Aldol de Aniones Ester y la Reacción de Reformatsky
La adición de un anión éster al grupo carbonilo de un aldehído o cetona es un tipo de adición aldólica (Sección 17-3):
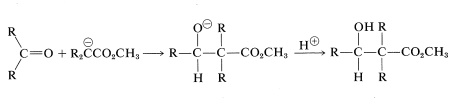
Existen ciertas dificultades para lograr este tipo de reacción aldólica. Primero, la hidrólisis de ésteres inducida por álcalis competiría con la adición. Segundo, podría intervenir una condensación de Claisen del éster, y tercero, el anión éster es una base más fuerte que los aniones enolato de aldehídos o cetonas, lo que significa que la reacción podría ser derrotada por transferencia de protones del tipo
Sin embargo, se puede lograr una reacción sintética útil de la siguiente manera. Primero, el anión éster se forma en ausencia de agua sin causar una condensación de Claisen u otra adición de carbonilo. Esto se puede hacer con etanoato de etilo tratándolo con bis (trimetilsilil) amida de litio en solución de oxaciclopentano en\(-80^\text{o}\):
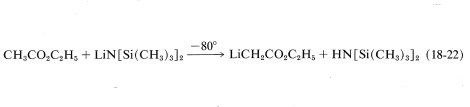
La ventaja de\(\ce{LiN[Si(CH_3)_3]_2}\) como base en esta reacción es que\(\overset{\ominus}{\ce{N}} \ce{[Si(CH_3)_3]_2}\) es una base razonablemente fuerte; es voluminosa, lo que inhibe la adición al carbonilo; y también forma una amina débilmente básica\(\ce{HN[Si(CH_3)_3]_2}\), que no interfiere en las reacciones posteriores.
La solución de litioetanoato de etilo debe mantenerse fría y tratarse puntualmente con un aldehído o cetona. Así, con 2-propanona,
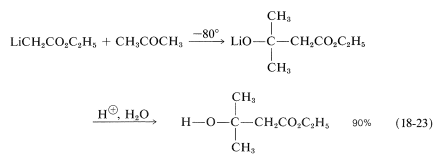
Para que la reacción sea exitosa, la adición de carbonilo tiene que ser más rápida que la reacción de transferencia de protones,\(\ce{LiCH_2CO_2C_2H_5} + \ce{CH_3COCH_3} \rightarrow \ce{CH_3CO_2C_2H_5} + \ce{LiCH_2COCH_3}\) y, en\(-80^\text{o}\), este es el caso. Esta síntesis de\(\beta\) -hidroxiésteres es un hermoso ejemplo de cómo se pueden manipular las tasas de reacciones competitivas para obtener un alto rendimiento de un producto de adición deseado que puede no ser el más termodinámicamente favorable.
Una síntesis estrechamente relacionada de\(\beta\) -hidroxiésteres es proporcionada por la reacción de Reformatsky. Esta síntesis comienza con un aldehído o cetona\(\ce{RCOR'}\), y un\(\alpha\) -bromo éster, como el bromoetanoato de etilo. El zinc en un disolvente no hidroxílico (generalmente benceno) transforma el éster de bromo en un compuesto de organozinc, que luego se agrega al aldehído o cetona carbonilo. La hidrólisis produce el\(\beta\) -hidroxi éster:
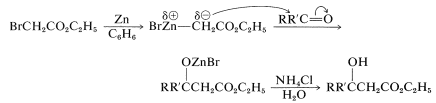
Al igual que los aldoles,\(\beta\) -hidroxiésteres se deshidratan (generalmente fácilmente) a\(\alpha\) compuestos carbonílicos\(\beta\) insaturados.

Condensaciones Biológicas de Claisen y Adiciones Aldólicas. Metabolismo de ácidos grasos
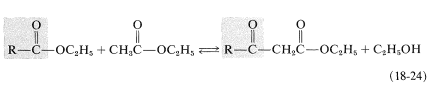
Los resultados globales de una condensación de Claisen es la transferencia de un grupo acilo\(\left( \ce{RCO}- \right)\) de una molécula de éster a otra:
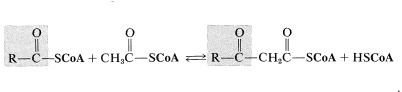
Lo contrario de la reacción anterior es un paso clave en la degradación oxidativa de los ácidos grasos. Esta condensación inversa de Claisen (catalizada por tiolasa) implica la escisión de un enlace carbono-carbono de un\(\beta\) -ceto-éster de la coenzima A por otra molécula de coenzima A para dar un nuevo derivado de acilo (\(\ce{RCO-S}\)CoA) y derivado de etanoilo (acetilo) (\(\ce{CH_3O-S}\)CoA) ):
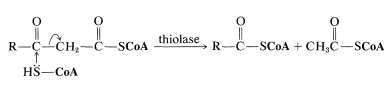
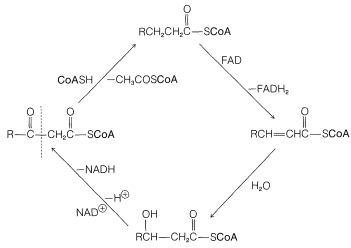
Después de la formación del\(\beta\) -cetotioéster, se escinde por CoA\(\ce{SH}\), y el tioéster resultante vuelve a la secuencia dos carbonos más cortos que antes. De esta manera, un ácido graso se degrada desde el extremo carboxilo, dos carbonos a la vez.
Existen dos vías principales para la utilización de la etanoil coenzima A (\(\ce{CH_3CO-S}\)CoA) formada en cada giro del ciclo de oxidación de la Figura 18-8. O bien se usa para sintetizar moléculas más grandes como ácidos grasos, esteroides, etc., como se describirá en la Sección 30-5A, o el grupo acilo se oxida a\(\ce{CO_2}\) y\(\ce{H_2O}\):
\(\ce{CH_3CO-S}\)CoA\(\overset{\left[ \ce{O} \right]}{\rightarrow} 2 \ce{CO_2} + \ce{H_2O} + \ce{HS}\) CoA
La oxidación del grupo acilo de la coenzima A es el resultado neto del ácido cítrico o ciclo de Krebs (Sección 20-10B). Aquí nos interesará el punto de entrada del ciclo mediante el cual se emplea la etanoil coenzima A en una reacción que construye la\(\ce{C_6}\) cadena de ácido cítrico (ácido 3-carboxi-3-hidroxipentanoico) a partir de\(\ce{C_2}\)\(\ce{C_4}\) piezas:
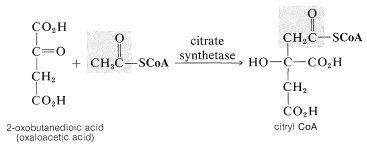
Esta reacción es bastante especial ya que es una adición de tipo aldol en la que un tioéster es el donador (nucleófilo) y un cetoácido es el aceptor (electrófilo). De la discusión en\ (\ alpha\) Carbones de derivados de ácidos carboxílicos” href=” /librerías/química_orgánica/libro:_básico_principios_de_química_orgánica_ (Roberts_and_caserio) /18:_carboxilic_ácidos_y_su_derivativos/18.09:_reacciones_at_el/ (/alpha/) _carbons_of_derivatives/18.09:_reacciones_at_the/ (/alpha/) _carbons_of_derivatives/Derivados de ácidos carboxílicos #18 -8E_Aldol -TYPE_ADDICIONES_OF_ESTER_ANIONS_AND_The_Reformatsky_reacción">Sección 18-8E, verá que reacciones de este tipo que involucran a un éster como donante y a un aldehído o cetona como aceptor se pueden lograr en el laboratorio solo bajo condiciones bastante especiales. Para que el tioéster funcione como nucleófilo en el\(\alpha\) carbono bajo las restricciones impuestas al hacer que la reacción ocurra al pH fisiológico, la enzima catalizadora debe promover casi con certeza la formación de la forma enol del tioéster. El enol entonces podría agregarse a la cetona carbonilo con la ayuda de un grupo básico en la enzima. Este tipo de catálisis por enzimas se discute en la Sección 25-9C.
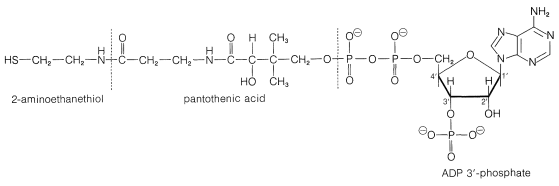
\(^5\)Es posible una confusión considerable debido a la forma en que los bioquímicos utilizan nombres abreviados y fórmulas para los derivados acilo de la coenzima A. Para enfatizar el\(\ce{-SH}\) grupo vital, la coenzima A suele escribirse como CoA\(\ce{SH}\). Sin embargo, los derivados de acilo con mayor frecuencia se llaman acetil CoA y similares, no acetil\(\ce{S}\) CoA, y bien podría tener la impresión errónea de que el azufre de alguna manera ha desaparecido en la formación del derivado de acilo. Incluiremos el azufre en fórmulas como\(\ce{CH_3COS}\) CoA, pero usaremos los nombres habituales como acetil CoA sin incluir el azufre. Para dejar claro que el CoA no contiene cobalto, el CoA se imprime en este texto en negrilla.
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."