6.1: Colchicina
- Page ID
- 70224

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Estrategia biosintética
El uso de aminoácidos aromáticos, es decir, fenilalanina o tirosina, como únicos materiales de partida para la colchicina (8) requiere preparar el anillo C de siete miembros por expansión de un anillo aromático de seis miembros en un precursor como el 9. La forma en que se logra la expansión del anillo se considerará más adelante. Dado que los aminoácidos de partida no tienen grupos amino bencílicos, es probable que el enlace entre el anillo C y el carbono bencílico esté formado por sustitución aromática electrófila. Dado que los aminoácidos de partida son derivados del ácido aril propiónico, el electrófilo podría derivarse de una amida aril propiónica tal como 10. Claramente, el enlace entre los anillos A y C en 10 se formaría por acoplamiento oxidativo de precursores de arilo ricos en electrones 11 y 12. Dado que el nitrógeno en 11 probablemente proviene de un grupo amino en el precursor de aminoácido del anillo C, y dado que 12 también debe derivarse de un ácido a-amino arilpropiónico, es probable que la estructura 12 deba revisarse para que el grupo R en 11 incorpore 12 como en la amida 13 (ver abajo). Un ácido cinámico 14, generado por la eliminación del amoníaco de la fenilalanina (6) podría ser el progenitor de la porción de ácido arilpropiónico de la amida 13.
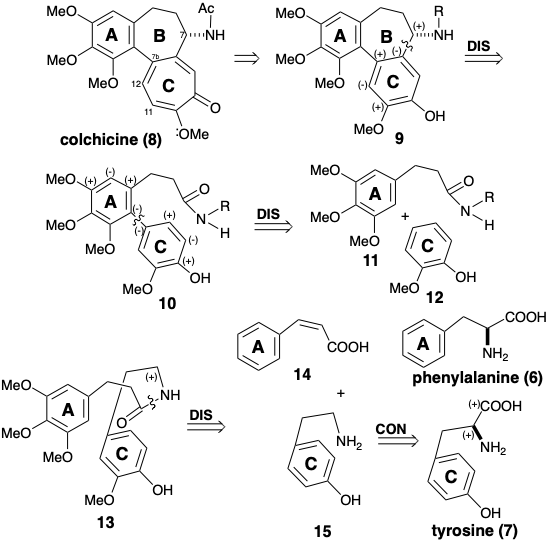
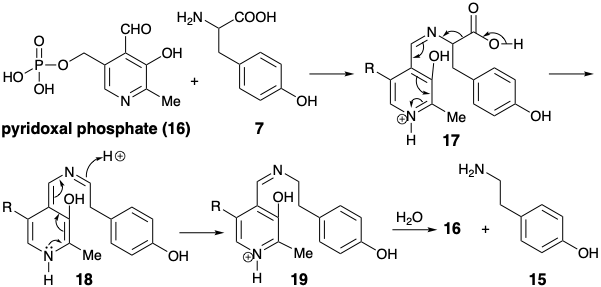
La generación de una feniletil amina 15 por descarboxilación de tirosina (7) requiere la escisión de un enlace que se encuentra en un circuito disonante. Tal escisión se logra biosintéticamente mediante un proceso polar que convierte el grupo amino temporalmente en un derivado que puede estabilizar el exceso electrónico en el carbono amino. La descarboxilación de tirosina (7) para dar p-hidroxi-b-fenilamina (15) es un ejemplo de una reacción general de aminoácidos que es promovida por la coenzima piridoxal fosfato (16). El proceso es iniciado por la formación de una base Schiff 17. El nitrógeno de la piridina se conjuga con el carbono a al carboxilo y puede estabilizar el exceso electrónico en ese carbono. El nitrógeno de la imina no proporciona activación polar; sirve meramente como un átomo de enlace. La base de Schiff 17 se somete fácilmente a descarboxilación catalizada por ácido para formar 18. La protonación de 181 conduce a la rearomatización entregando la base Schiff 19. La hidrólisis entrega la feniletil amina 15 y regenera 16. Así, el fosfato de piridoxal (16) actúa como catalizador de inversión de la reactividad polar.
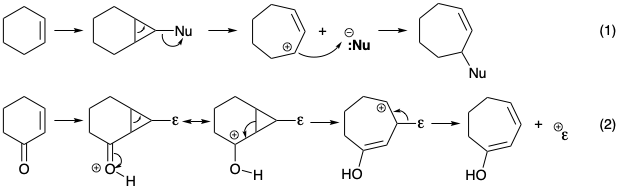
Se encontraron estrategias de expansión de anillos polares en la síntesis total de longifoleno (ver sección 4.4). Allí, un intermedio bicíclico fusionado, generado por ciclopropanación de un ciclohexeno, se sometió a escisión del enlace de fusión junto con la salida de un nucleófugo para suministrar una matriz de cicloheptenilo (eq 1). Un proceso análogo, que implica la salida de una electrofuga junto con la escisión del enlace de fusión en un intermedio similar, depende de un reordenamiento ciclopropil carbinilo a carbocatión homoalilo (eq 2). La presencia de sustituyentes de oxígeno en el anillo C de colchicina sugiere la posibilidad de que dicho mecanismo de expansión de anillo pueda producirse durante la conversión biosintética de un precursor de anillo C de seis miembros en el anillo C de siete miembros.
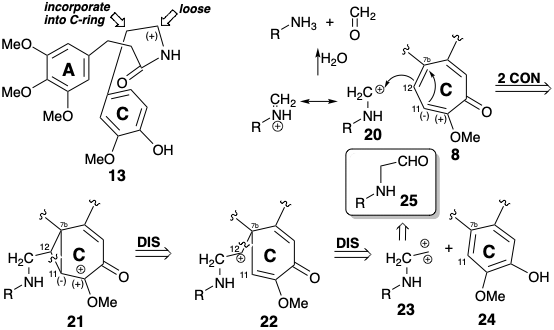
Un carbono de la cadena lateral de fenetilo en 13, el carbono bencílico, podría incorporarse en un precursor aromático para generar el anillo C de siete miembros. El carbono restante de la cadena lateral debe estar desconectado. La desconexión de este carbono como carbocatión puede ser estabilizada por el grupo amino como en 20. El análisis retrosintético de la expansión del anillo biosintético de colchicina, presumiendo 20 como electrofuga, sugiere los intermedios de carbocatión ciclopropil carbinilo y homoalilo 21 y 22. El carbono que se inserta en el anillo aromático del material de partida 24 corresponde al sintón de dicación 23 para el cual un aldehído podría servir como equivalente sintético.
Biosíntesis
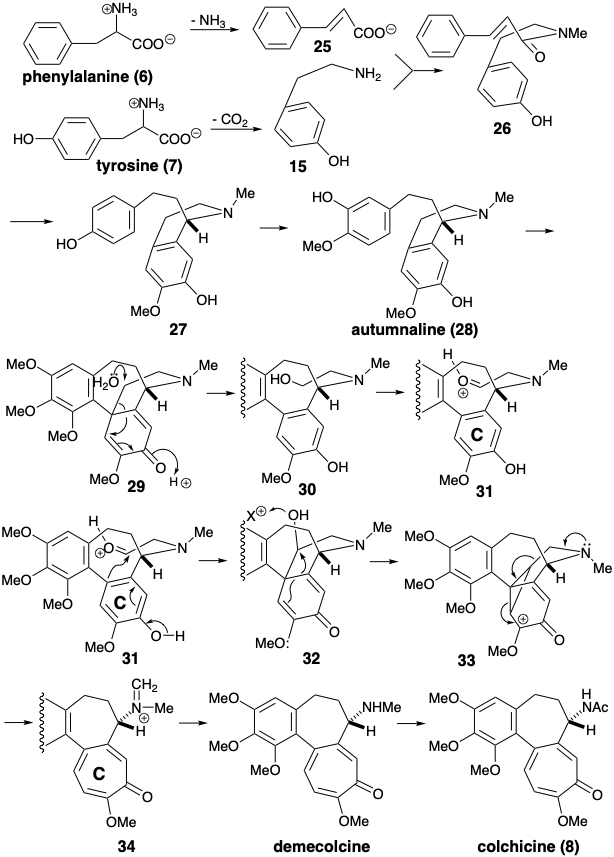
En la biosíntesis de la colchicina (8),\(\ce{NH3}\) se elimina de la fenilalanina (6), y se descarboxila la tirosina (7), antes de que se forme una amida 26 uniendo los intermedios resultantes 25 y 15. El anillo B de siete miembros es creado por una sustitución aromática electrofílica intramolecular enantioselectiva que da 27 y un acoplamiento oxidativo de 28 que entrega 29. La funcionalización de un metileno aparentemente inactivado en 29 se logra mediante una secuencia que implica fragmentación hidrolítica polar a 30 seguida de oxidación a un aldehído adecuado para inserción en el progenitor del anillo C de seis miembros en 31. La expansión del anillo arilo para generar una tropolona de siete eslabones se inicia por sustitución aromática electrófila intramolecular. La alquilación intramolecular de 32 seguida de una fragmentación de un ciclopropano intermedio 33 produce el esqueleto expandido de anillo en 34 de la diana biosintética. La hidrólisis final del grupo imminium, la N-desmetilación y la N-acetilación liberan colchicina (8).
Características Moleculares
La estabilidad de los derivados aromáticos a menudo se explota en síntesis mediante estrategias que incorporan restos aromáticos preformados. Así, en la biosíntesis de la colchicina (8), el anillo A aromático se deriva del anillo aromático preformado de la fenilalanina (6). Las cuatro síntesis totales diferentes de colchicina a considerar en esta sección adoptan esta misma estrategia. En contraste con la biosíntesis, sin embargo, todas las síntesis totales emplean materiales de partida aromáticos completamente funcionalizados. Esto se debe a que las hidroxilaciones regioselectivas, que se logran enzimáticamente en la biosíntesis, no se logran tan fácilmente en el laboratorio.
También hay que señalar que el anillo C de la colchicina (8) contiene dos grupos funcionales que proporcionan activación electrófila en átomos de carbono adyacentes, una disonancia de reactividad polar. Así, estos grupos funcionales no pueden ser explotados directamente en una reacción polar (es decir, sin umpölung) para crear el enlace C-C del circuito disonante entre estos átomos de carbono. En cada una de las siguientes síntesis, el anillo C de siete elementos se agrega a un precursor de anillo AB. En cada caso, se explota una estrategia diferente para la anulación del anillo C.
Estrategias clave dirigidas a intermediarios para la conchicina
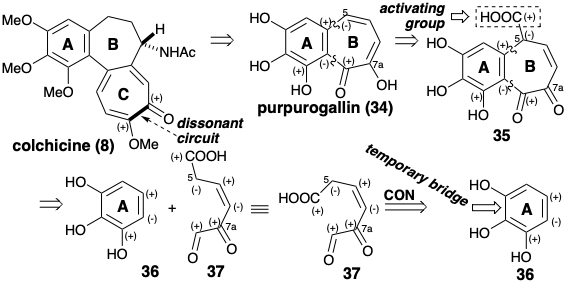
Dos síntesis de colchicina aprovecharon un intermedio clave fácilmente disponible, purpurogalina (34), para el grupo de anillo AB de 8 y formaron el anillo C de siete miembros por reacciones no polares. El análisis de reactividad polar de 34 sugiere una síntesis a partir de un precursor aromático 36. Así, el apéndice a 36 del anillo B en 34 se puede facilitar mediante la adición de un carboxilo activador como en 35 que podría generarse a partir del 36 y 37 por dos reacciones formadoras de enlaces polares. De hecho, el material de partida aromático 36 también puede ser el precursor de un equivalente sintético 39 temporalmente puenteado (vide infra) del sintón 37.
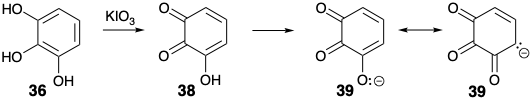
La purpurogalina (34) fue un producto bien conocido de la oxidación del pirogalol (36). Probablemente se forma por dimerización de 3-hidroxi-o-quinona (38). Una adición inicial de Michael del enolato 39 a 38 para dar 40 seguido de una condensación intramolecular de aldol conduce a un intermedio tricíclico 41 (ver más abajo). Esto se escinde en una reacción retro Dieckman, típica de β-dicetonas, para producir un ácido carboxílico bicíclico 37, que puede aislarse. Este β-ceto-ácido vinilógico se somete fácilmente a descarboxilación para suministrar 28.
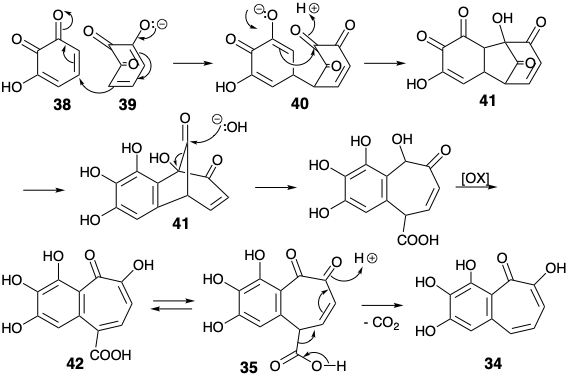
Ambas síntesis empleando 34 para los anillos A y B simplificaron el objetivo al descuidar el grupo acetamido. Esto podría introducirse por oxidación bencílica después de completar el esqueleto de carbono de la diana 8. Ambas síntesis construyen la diana simplificada 43 a partir del derivado de benzosuberona 44, en el que el grupo funcional carbonilo proporciona activación para la anelación del anillo C. Eschenmosher 1 preparó 39a reduciendo el trimetil éter 45 de purpurogalina (34) vía 46, 47 y 48.
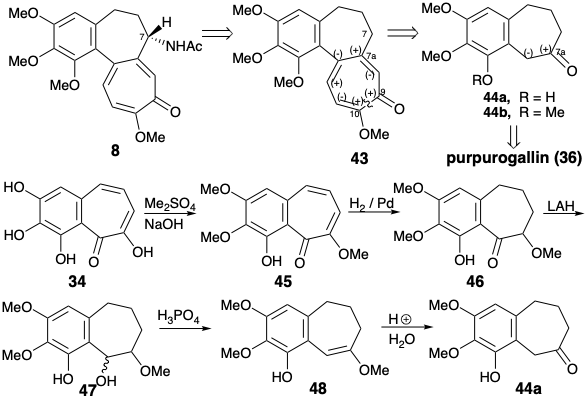
Una estrategia de reordenamiento cicloadición-pericíclico para el anillo C
La estrategia 1 de Eschenmosher para la anelación del anillo C de siete miembros fue construir un dieno sobre 44a, luego construir un carbociclo de seis miembros mediante una reacción de Diels-Alder del dieno y finalmente expandir el anillo de seis a siete miembros mediante reordenamiento pericíclico de un norcaradieno. La fácil interconversión de cicloheptatrienos como 49 con norcaradienos como 50 es un reordenamiento sigmatropico (Cope) bien conocido [3.3] que es impulsado a favorecer a los cicloheptatrienos por el alivio de la cepa de anillo asociada con la escisión del ciclopropano. Es dudoso que la estrategia fuera concebida por rigurosos análisis retrosintéticos ya que la conversión de 49 a 43 sin duda requeriría extensas manipulaciones de grupos funcionales. La decisión de emplear 49 como precursor para 43 casi con certeza evolucionó como consecuencia de las decisiones de emplear: (1) un reordenamiento Cope de un norcaradieno.
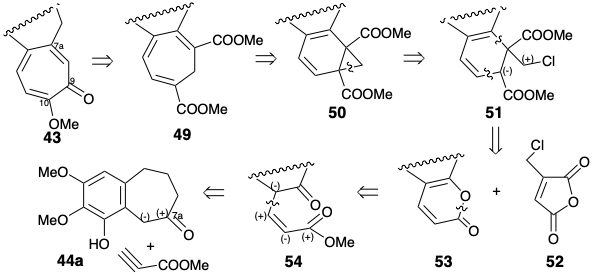
Por lo tanto, el anhídrido clorometilmaleico (52) es ideal para cicloañadir al dieno relativamente rico en electrones 53. Quizás una dislocación más obvia de 50 sería a 53 y el ciclopropeno 55. Esta rama del árbol retrosintético, muy probablemente, se consideraría primero porque proporcionaría una síntesis más convergente. Así, la reacción de 55 con α-pirona 53 entregaría 50 directamente por una cicloadición de aliso Diels, seguida in situ por una cicloeliminación retro de Diels Alder de dióxido de carbono de un intermedio 56.
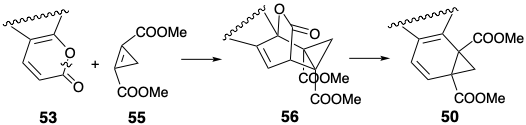
El precursor alternativo, el dienófilo 52 con un sustituyente clorometilo, se sugiere por desconexión polar de 50 a 51 que puede derivarse de 53 por una secuencia Diels-Alder-retro Diels-Alder. Se eligió el dienófilo 52 en lugar de 55 porque está más fácilmente disponible que 55. Una adición de Diels-Alder de 52 a 53 seguida de una eliminación retro Diels-Alder\(\ce{CO2}\) de un aducto intermedio, un proceso de intercambio carbonil-alqueno, proporcionará el ciclohexadieno 51. La fuerza impulsora para el proceso es la generación de un enlace C=O relativamente estable a cambio del enlace C=C del dienófilo 52. El dieno 53 es una enol lactona derivable del ácido 54. El análisis polar de 54 sugiere la construcción a partir de 44a y propiolato de metilo mediante una adición polar de 1,4-adición. La aparente alquilación de Michael de 44a con propiolato de metilo proporcionó pirona 57. Aunque 44a podría dar 57 vía directa Michael adición a la yneona, de hecho la reacción fue más compleja. Implicaron la participación del anión fenolato. Así, el electrófilo se suministró intramolecularmente al carbono bencílico bastante impedido de 44a.
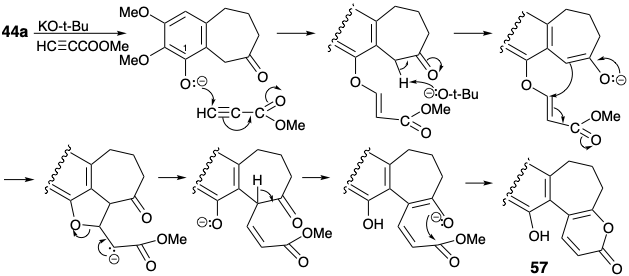
Después de la metilación del fenol 57, la anelación de un ciclohexadieno 58 se logró mediante la conocida reacción de cicloadición-cicloeliminación de α-pironas. Alquilación intramolecular catalizada por bases del diéster 51 de 58 led, a través del norcaradieno 50, al cicloheptatrieno 49. El éster menos impedido en 49 fue fácilmente hidrolizado selectivamente, y el ácido resultante proporcionó una tropolona 61 a través de hidroxilación-descarboxilación vecinal catalizada por osmio, saponificación del éster restante en 60 y una segunda descarboxilación.
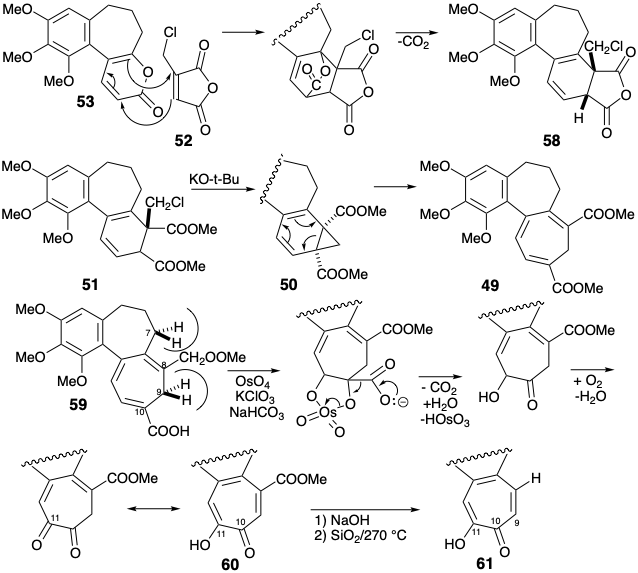
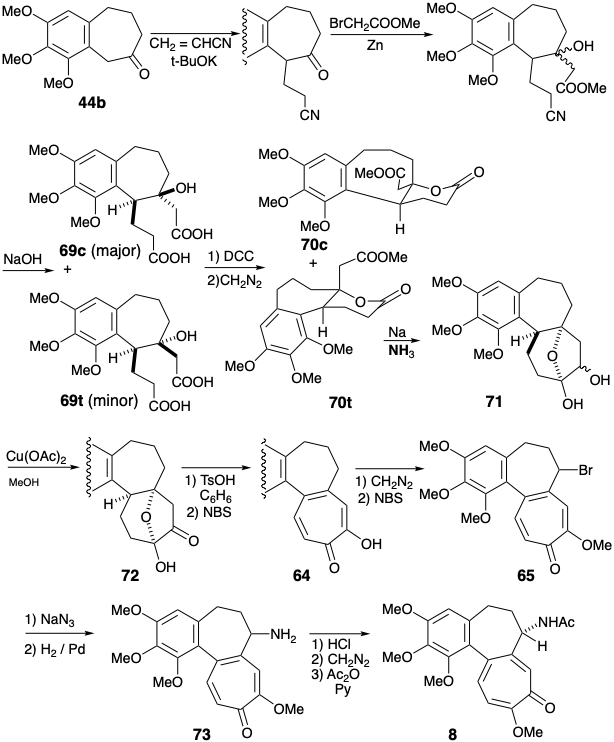
Desafortunadamente, la secuencia conduce a una función de oxígeno en C-11 en lugar de C-9 como se requiere para la colchicina. La transposición del grupo funcional se logró mediante una secuencia bien precedente de desplazamientos nucleofílicos sobre el derivado de tosilato 62 de 61 primero con\(\ce{NH3}\) para dar 63 luego con -OH para entregar 64. La tropolona 64 se metiló y funcionalizó en C-7 mediante bromación alílica con N-bromosuccinimida para proporcionar 65. El desplazamiento nucleofílico de bromuro por amoníaco dio la amina C-7 requerida acompañada de amonólisis de la tropolona, un éster vinilógico. La saponificación de la amida vinílogosa resultante 66 dio 67, que proporcionó colchicina tras la metilación y acetilación.
Una estrategia de aciloína para el anillo C
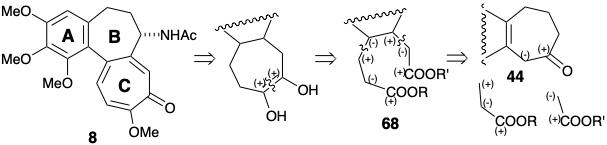
La estrategia de Van Tamelen para la anulación del anillo C con funcionalidad de oxígeno vecinal reconoce la aplicabilidad de una reacción de aciloína intramolecular para la creación del circuito disonante entre grupos activadores electrófilos vecinales. 2 El análisis polar adicional sugiere una síntesis del diéster intermedio 68 requerido mediante la explotación de la activación polar proporcionada por un grupo carbonilo en un precursor 44 del anillo AB. Así, el apéndice de las cadenas laterales de ácido acético y propiónico debería ser factible respectivamente por una reacción de Reformatsky y alquilación de Michael.
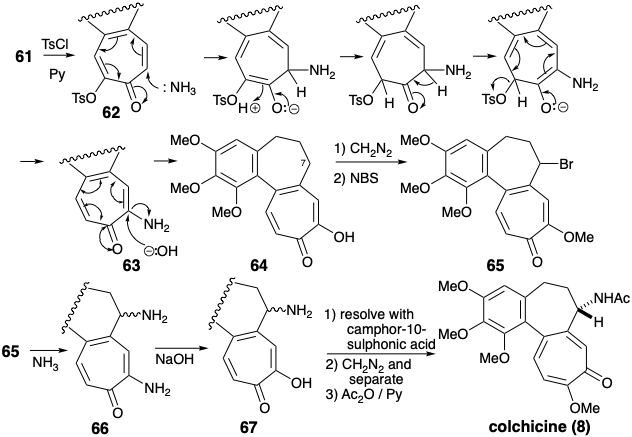
Se obtuvo un par de hidroxidiácidos isoméricos 69c y 69t por alquilación Michael de 44b con acrilonitrilo, reacción Reformatsky del cetonitrilo intermedio e hidrólisis. El grupo hidroxilo se enmascaró intramolecularmente como lactona, y el resto del grupo carboxilo fue metilado. Solo uno de los ésteres isoméricos de lactona 70 sufrió una reacción de aciloína que proporcionó 71. Desafortunadamente, este era el isómero menor 70t, con un sustituyente carbometoximetilo axial. Los grupos éster en el isómero principal 70c no pudieron alcanzar fácilmente la yuxtaposición adecuada para la reacción de aciloína intramolecular. El producto de aciloína 71 se oxidó a 72 con Cu (II) y se oxidó adicionalmente con NBS para proporcionar tropolona 64. La metilación y bromación liberaron el bromuro 65, un intermedio también preparado por Eschenmosher. La sustitución de un grupo amino por el sustituyente bromo en 65 seguida de hidrólisis, remetilación del ácido carboxílico vinilógico y N-acetilación produjo colchicina 8.
Se requirieron extensas manipulaciones de grupos funcionales después de completar el esqueleto de carbono en la síntesis de Eschenmosher debido a que se adoptó una estrategia de anulación para el anillo C que ignoró la funcionalidad relacionada con la diana. Van Tamelen pudo completar su síntesis con menos manipulaciones de grupos funcionales porque una mayor funcionalidad relacionada con el objetivo, que había sido explotada para facilitar la construcción esquelética, estaba presente después de la finalización del anillo C.
Una estrategia de formación de enlaces polares promovida por la funcionalidad relacionada con el objetivo
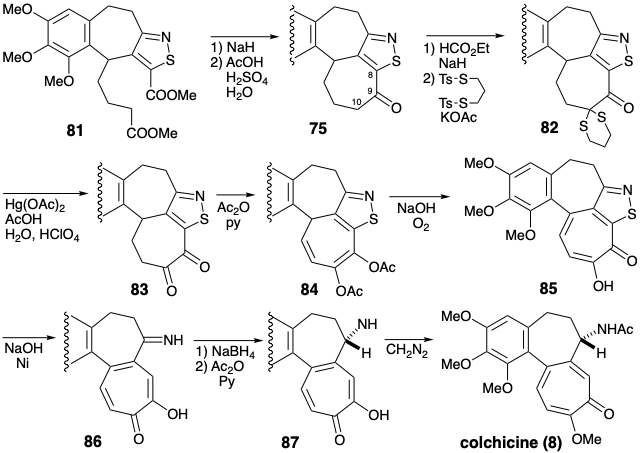
Otra síntesis de colchicina (8), que también implica la anelación del anillo C en un intermedio preformado del anillo AB, fue ideada por R. B. Woodward. 3 La estrategia es única en la incorporación temprana del sustituyente 7-amino y en su uso extensivo de la funcionalidad relacionada con la diana para facilitar la construcción esquelética de carbono. La dislocación de la diana 8 al derivado 74 en el que también se bloquea la posición 8 permite la introducción selectiva de oxígeno en C-10. En el equivalente sintético 75 del sintón 74, un anillo de isotiazol aromático enmascara tanto el sustituyente amino como el C-8. La anulación del anillo C se puede lograr mediante una ciclación Dieckman 77 → 76 explotando la activación polar proporcionada por el carbonilo C-9 y un grupo carbometoxilo activador unido a C-10 en un precursor 77. La activación polar proporcionada por este grupo carbometilo en 77 sugiere una sustitución aromática electrófila 78 → 77 para la anelación del anillo B. Por supuesto, para que se exprese la electrofilicidad apropiada, el grupo carbometoxilo en 77 debe conjugarse con la posición γ como en 78. El isotiazol también sirve como puente temporal en 78, que ayuda entropicamente en la ciclación 78 → 77. El carbometoxilo en 78 también permite una elaboración polar de la matriz de ésteres dienoicos a partir de un precursor de isotiazol aldehído 79 estabilizando un carbanión en el fragmento iluro 80. El átomo de azufre en el anillo de isotiazol incluso proporciona activación para la generación de carbaniones a azufre en 78 permitiendo la conexión polar del carbometoxilo requerido en 77. La amplia utilización estratégica de la unidad de isotiazol en la estrategia de Woodward es un sello distintivo de este plan sintético.

Después de la ciclación Dieckman de 81, la monocetona C-9 75 se oxida selectivamente en el C-10 mediante una reacción polar que explota la activación nucleofílica en C-10 proporcionada por el grupo carbonilo C-9. El ataque nucleofílico por el carbanión C-10 sobre un electrófilo de azufre conduce a la oxidación de C-10 (y reducción concomitante de azufre). El tiocetal resultante 82 se hidroliza a 83, lo que proporciona el producto de acetilación de enol 84. El enediolato obtenido por saponificación del diacetato 84 se oxida fácilmente para suministrar 85. La desulfuración de 85 con níquel Raney elimina el grupo enmascarante de isotiazol. La reducción de la imina resultante 86 seguida de acetilación proporciona 87 que se N-metila para suministrar colchicina (8).
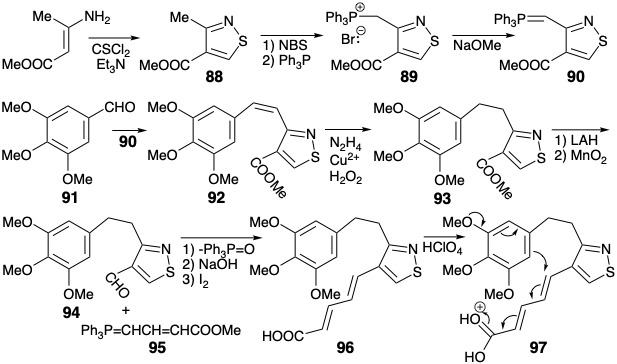
La síntesis de Woodward del intermedio clave 81 se centra alrededor del nuevo anillo aromático de isotiazol. El material de partida, un isotiazol 88, está fácilmente disponible a partir del β-aminocrotonato de metilo, una enamina derivada del acetoacetato de metilo. El grupo metilo conjugado en 88 se bromea fácilmente con NBS. La alquilación\(\ce{Ph3P}\) da un bromuro de fosfonio 89, que da iluro 90 tras la desprotonación. La olefinación de Wittig de 3,4,5-trimetoxibenzaldehído (91) con iluro 90 produce alqueno 92 que se hidrogenó selectivamente con diimida. La hidrogenación catalítica de 92 se vio imposibilitada por la susceptibilidad del anillo isotiazol a la hidrogenólisis. La reducción de hidruro del éster 93 y la oxidación parcial de un carbinol conjugado intermedio dieron el aldehído 94. La olefinación de Wittig de 94 con iluro 95 seguida de saponificación e isomerización cis-trans proporcionó 96.
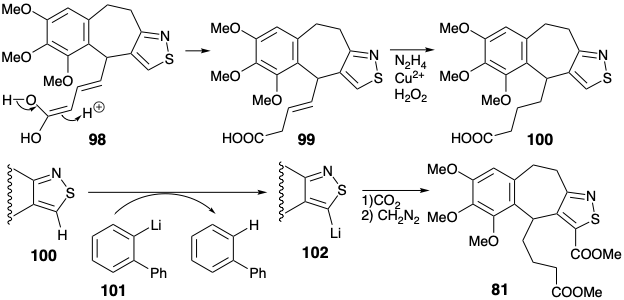
Este dieno se sometió a sustitución aromática electrofílica intramolecular tras el tratamiento con ácido perclórico. El puente temporal proporcionado por el anillo de isotiazol sin duda facilita esta ciclación al yuxtaponer favorablemente los centros de carbono reaccionantes. La reducción selectiva del producto de ciclación 99 con diimida dio 100. El carbono final requerido para el anillo C se introdujo por carbonilación de un derivado de organolitio 102 del tiazol 100. Así, la metalación selectiva en presencia de un carboxilato se logró con el aril litio 101, relativamente no nucleofílico, estéricamente gravado. El tiazol litiado 102 dio el intermedio clave 81 tras la carbonatación seguido de metilación.
Una estrategia hipotéticamente biomimética
Una cuarta síntesis de colchicina, ideada por Scott 4, difiere de las tres anteriores en su estrategia para la construcción esquelética. Scott ensambló un intermedio que contenía los anillos A y C y luego creó el anillo B mediante un acoplamiento oxidativo intramolecular. Esta estrategia se basó en un mecanismo hipotético para la biosíntesis de colchicina, que ahora se sabe que no es operativo. En este mecanismo, en contraste con el mecanismo biosintético real (ver arriba), la generación de un anillo C de tropolona por expansión de anillo de un precursor aromático precede al acoplamiento oxidativo que crea el anillo B.
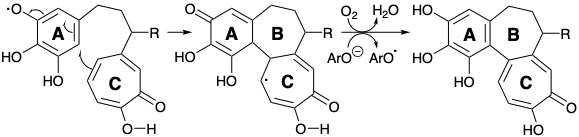
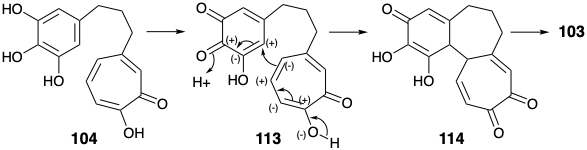
La táctica de introducir el grupo amino colchicina al final de la síntesis tuvo un buen antecedente. Así, la estrategia de Scott comienza con la dislocación del objetivo 8 a un precursor 103. El enlace entre los anillos arilo y tropolona se encuentra a lo largo de un circuito disonante entre la funcionalidad oxígeno en las posiciones 1 + 9, 1 + 10, 3 + 9 o 3 + 10. Así, el análisis polar revela que la formación de este enlace no se puede lograr mediante un proceso polar utilizando estos grupos funcionales para proporcionar activación ya que se requeriría la unión de dos centros nucleofílicos. Tal proceso se puede lograr oxidativamente, sugiriendo dislocación de 103 a un precursor 104 con dos monocíclicos unidos por un simple puente trimetileno. La reactividad polar proporcionada por el grupo carbonilo en 104 puede reforzarse activando grupos carboxilo en un precursor 105 permitiendo la formación de enlaces polares entre un intermedio electrófilo 106 y un nucleófilo derivado de 107 por desprotonación. La elección no obvia de 107 como precursor para 104 fue dictada sin duda por su fácil disponibilidad a partir de purpurogalina 34 por escisión oxidativa selectiva del anillo arilo relativamente rico en electrones.
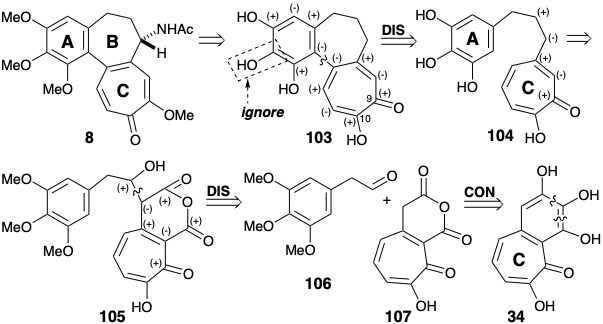
Así, la oxidación de 34 con peróxido de hidrógeno seguida de deshidratación dio el anhídrido enol 107. Es interesante que Scott utilizó purpurogalina (34) como precursor del anillo C de la colchicina en contraste con Eschenmosher y Van Tamelen quienes construyeron los anillos A y B de la colchicina a partir del 34.
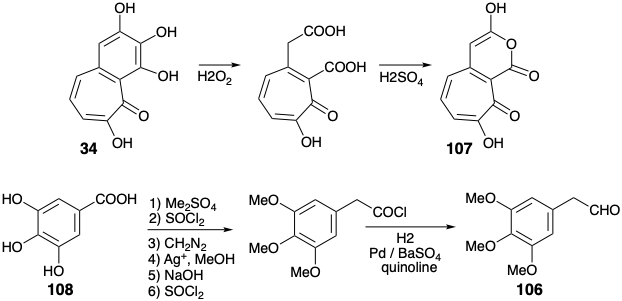
Se obtuvo un sintón de anillo A, 3,4,5-trimetoxi acetaldehído (106) por homologación Arndt-Eistert de ácido 3,4,5-trihidroxibenzoico (108). La unión del sintón de anillo A electrófilo 106 con el sintón nucleofílico de anillo C 107 se logró mejor térmicamente sin catálisis base mediante un proceso que comienza con una condensación aldólica. A 100°C, se formó una lactona 110, presumiblemente por descarboxilación de un β-ceto-ácido vinilógico intermedio 109. El calentamiento adicional de 110 a 190°C dio 112, presumiblemente por descarboxilación de un intermedio β-ceto-ácido 111. La reducción y desmetilación dieron el anillo A+C intermedio 104. El producto rico en electrones 103 del acoplamiento oxidativo deseado de 104 fue altamente susceptible a una oxidación adicional indeseable. Sin embargo, el oxidante suave\(\ce{FeCl3}\), en un sistema bifásico dio 103 después de la cromatografía en papel en atmósfera inerte aunque con bajo rendimiento (5%).
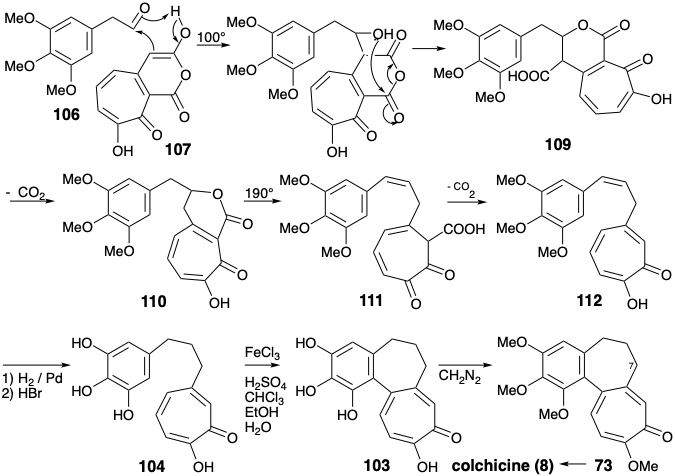
Se ha sugerido que la anulación puede implicar un proceso iónico en lugar del acoplamiento radical originalmente previsto. 5 Así, utilizando la funcionalidad aril oxígeno en la posición 2, que fue ignorada en el análisis polar de 103 anterior, se puede observar que el enlace entre los anillos arilo y tropolona se encuentra en un circuito consonante entre las posiciones 2 y 10. Esto permite la adición de Michael de un nucleófilo de tropolona a un electrófilo de enona como se muestra en 113 para suministrar 114. El ajuste final de la funcionalidad dio 8 de 103 a 73 como se discutió anteriormente.