
1.4: Ácidos y bases de Lewis, electrófilos y nucleófilos




- Última actualización
- 30 oct 2022
- Guardar como PDF

( \newcommand{\kernel}{\mathrm{null}\,}\)
Como hemos visto, cualquier reacción en la que un protón (H+) se transfiera de una molécula a otra puede considerarse como una reacción ácido-base de Lewis, pero ahora es el momento de ampliar el alcance de las reacciones ácido-base de Lewis. El requisito estructural para una base de Lewis es esencialmente el mismo que los que discutimos para una base Brønsted. Es decir, una base de Lewis debe tener un par de electrones solitarios accesibles que puedan ser donados en un enlace con un ácido de Lewis. Por ejemplo, muchas (pero no todas) moléculas que contienen nitrógeno y oxígeno tienen tales pares de electrones solitarios disponibles y por lo tanto pueden considerarse como bases de Lewis. [12] Es el ácido de Lewis el que puede tomar varias formas diferentes (y por lo tanto, puede ser más difícil de reconocer). Un ácido de Lewis debe ser capaz de aceptar un par de electrones. En la práctica esto significa que una variedad de sustancias (ademásH+) pueden actuar como ácidos de Lewis: por ejemplo, cualquier especie con orbitales vacíos que sean energéticamente accesibles puede ser un ácido de Lewis. Ejemplos comunes de esta situación son compuestos de elementos del Grupo III (específicamenteB yAl); estos tienen sólo tres electrones de valencia. Los ejemplos incluyenBF3 (↑) yAlCl3, [13] ambos tienen una carga positiva parcial en el átomo central y un orbital vacío que puede aceptar electrones.
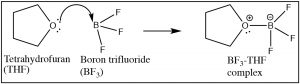
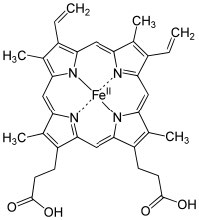
Otros ejemplos de ácidos de Lewis son iones metálicos comoFe2+,Fe3+,Cu2+, yMg2+; estos, por definición, tienen orbitales vacíos. La misma situación es cierto para muchas sales de metales de transición, por ejemploTiCl4 yNiCl2. [14] En los sistemas biológicos, los ejemplos de complejos ácido-base de Lewis incluyen el sitio activo del complejo de transporte de oxígeno en la hemoglobina (y la mioglobina), que consiste en un ion hierro complejado con 4 nitrógenos, los cuales forman parte de un anillo de porfirina. Un complejo similar de hierro-porfirina se encuentra asociado con las proteínas del citocromo que participan en la reacción de síntesis de ATP asociada en la cadena de transporte de electrones de las mitocondrias (→). La clorofila, el pigmento verde que forma parte del sistema de captura de luz en algas y plantas tiene una estructura similar, excepto que el ácido de Lewis en el centro del complejo esMg2+ más queFe2+. Esto tiene el interesante efecto de hacer que las especies de clorofila parezcan verdes, en lugar del rojo observado en la sangre. Esto es causado por la diferencia de brechas de energía entre los orbitales moleculares en unFe complejo en comparación con unMg complejo con un anillo de porfirina. Discutiremos este efecto con más detalle más adelante. Como veremos más adelante, los ácidos de Lewis son una clase importante de reactivos en química orgánica porque pueden interactuar con una amplia gama de bases.
Electrofilos y Nucleófilos
El siguiente paso lógico para expandir nuestras ideas sobre los ácidos y bases de Lewis es considerar reacciones que involucren carbono. Primero consideraremos reacciones en las que el carbono actúa como el ácido de Lewis, es decir, acepta un par de electrones para formar un nuevo enlace con una base de Lewis. Entonces, ¿qué situaciones haríamos un acto de carbono de esta manera? Podemos descartar (por ahora) compuestos de carbono con un orbital vacío (similar al boro). ¿Por qué? Porque todos los compuestos de carbono estables forman 4 enlaces y no hay orbitales vacíos bajos que puedan usarse para aceptar electrones.
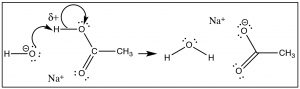
Pero veamos primero la reacción de transferencia de protones (H+) como modelo (→). En este caso el enlace con la base de Lewis (OH−) se forma al mismo tiempo que se rompe el enlace a la base conjugada (del ácido). Vemos que elδ+ on losH medios que el enlace con elH está parcialmente ionizado. ElH está “en camino” de convertirse enH+ una especie que sí tiene una órbita vacía y accesible. Elδ+ on theH atrae la carga negativa (oδ−) sobre la base, y se inicia la reacción, formando un nuevo enlace entre elO y elH, y al mismo tiempo rompiendo elO−H vínculo antiguo.
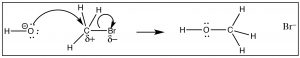
Podemos imaginar que un compuesto de carbono con unδ+ on theC podría comportarse de una manera muy similar. En esta molécula (H3CBr) elC−Br enlace se polariza de manera que hay una pequeña carga positiva en elC, que atrae al hidróxido cargado negativamente (→). La formación delO−C enlace ocurre con la rotura simultánea delC−Br enlace.
Consideremos las analogías entre estas dos reacciones —los mecanismos de cómo y por qué se mueven los electrones son similares. La única diferencia real entre las dos reacciones es que en la primera unaH+ se transfiere de unC (sobre el ácido carboxílico) a laOH−, mientras que en la segunda, se transfiere un grupo metilo a laOH−. Ahora, para un cambio de nomenclatura: cuando tal reacción involucra unC átomo (un centro de carbono) en lugar de llamar al carbono deficiente en electrones un ácido de Lewis, lo llamamos electrófilo (amante de electrones o carga negativa). De manera similar, el ion hidróxido (que actúa como una base de Lewis) ahora se llama nucleófilo (amante de la carga positiva). ¡Este cambio de terminología no es solo para confundir a los estudiantes! De hecho, existen diferencias sutiles entre ácidos y bases de Lewis y electrófilos y nucleófilos que hacen útil la distinción entre los dos. En particular, si bien todas las bases de Lewis son nucleófilos, como veremos, no todos los nucleófilos son bases.
Entonces ahora tenemos que preguntarnos, ¿qué factores hacen de un particularC dentro de una molécula un electrófilo? ¿Cómo podemos reconocer a un nucleófilo? ¿Qué criterios utilizamos para estimar la fuerza de un electrófilo o nucleófilo en particular? ¿Alguna vez podremos conseguir que el carbono actúe como nucleófilo? Si podemos responder a estas preguntas, podemos predecir el resultado de una amplia gama de reacciones.
¿Qué hace que un carbono en particular sea un electrófilo?
Los compuestos orgánicos más simples son los hidrocarburos, y los más simples de los hidrocarburos se conocen como alcanos. Los alcanos suelen tener la fórmulaCnH2n+2 (oCnH2n si hay un anillo de carbonos, restar2H por cada anillo extra). Todos los enlaces dentro de un alcano son enlaces sigma (sencillos); no contienen enlaces pi (dobles). [15] En un alcano, cada carbono está completamente saturado, hace cuatro enlaces simples y (como se señaló anteriormente) no hay dobles o triples enlaces. C−Clos enlaces son por supuesto, completamente no polares ya que los electrones están igualmente distribuidos entre dos átomos idénticos, sin embargoC−H los enlaces también son relativamente no polares ya que las electronegatividades deC yH son bastante similares. En la práctica esto significa que los alcanos son limitados en su reactividad. Las reacciones más comunes en las que un alcano puede tomar parte son reacciones con oxígeno para producirCO2 yH2O. Esta reacción es altamente exotérmica, aunque existe una energía de activación significativa, por lo que requiere una entrada inicial de energía (típicamente una chispa, un fósforo ardiente) para iniciar la reacción, pero luego la energía de la formación de los fuertesC=O yO−H enlaces (razón por la cual la reacción es exotérmico) se puede utilizar para iniciar más reacción. El mecanismo de reacción real es complejo; procede a través de una serie de radicales libres altamente reactivos (inestables) (especies con electrones desapareados) [16]. Si bien esta reacción es obviamente muy importante, así es como generamos gran parte de la energía para hacer funcionar nuestros autos y centrales eléctricas, desde una perspectiva de química orgánica no es muy interesante en gran parte porque es más o menos incontrolable. Es decir, si tienes suficiente oxígeno una vez iniciada la reacción generaCO2 yH2O, independientemente del hidrocarburo con el que comiences. [17]
Todo esto es para decir que los alcanos no son buenos candidatos para el tipo de reacciones que estamos considerando, no tienen ni carbonos nucleófilos ni electrófilos. Entonces, volvamos nuestra atención a los compuestos de carbono con elementos distintos deC yH y tanto enlaces sigma como pi (esto es, por supuesto, el resto de la química orgánica). Aquí encontramos una situación muy diferente: la gama de reacciones y los tipos de productos pueden parecer casi ilimitados. Si bien es imposible (y ciertamente indeseable) memorizar cada reacción y cada producto potencial, es posible organizar su comprensión de los sistemas químicos para que pueda hacer predicciones plausibles sobre qué reacciones pueden ocurrir. Al conocer los mecanismos de reacción, y cuándo son relevantes, también se puede predecir qué reacciones ocurrirán y por lo tanto qué productos se formarán. Como puede reconocer, esta es la misma estrategia que hemos utilizado para considerar las reacciones ácido-base, que se pueden entender mucho más ampliamente que las reacciones simples de transferencia de protones (H+). Pensar en un contexto electrófilo-nucleófilo proporciona una entrada en gran parte de la química orgánica.
Para que ocurran reacciones (distintas de las reacciones que involucran radicales libres, como la combustión), generalmente hay un “mango” dentro del sustrato: un lugar donde la densidad electrónica no se distribuye uniformemente, un sitio en el que los reactivos de carga opuesta interactúan (y reaccionan). En el ejemplo que usamos anteriormente, el carbono electrófilo tiene unδ+ sobre él; esta carga parcial surgió porque elC estaba unido a un elemento más electronegativo. Tal carga parcialmente positivaC es atractiva para cualquier especie con una carga negativa (o parcial negativa). Nótese que, por ahora, vamos a restringir el tipo de átomo de carbono que estamos considerando ya sea a un primario (es decir, un carbono con solo un grupo alquilo (denotado porR) y 2 hidrógenos,CH2R−) o un carbono metílico (CH3−). Como veremos, las cosas se vuelven más complicadas cuando comencemos a agregar más grupos alquilo alrededor del sitio del ataque, así volveremos a eso más tarde.
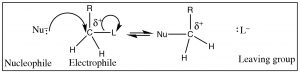
Para identificar a tal carga parcialmente positivaC se buscaríanC's unidos a grupos (átomos) que son más electronegativos, es decir, que actuarán para retirar electrones del carbono (denotado porL abajo). Pero como el carbono no puede formar más de cuatro enlaces ya que el nucleófilo entra y forma un enlace, otro enlace debe romperse. El átomo electronegativo (L) (o grupo de átomos), se conoce como el “grupo de salida” (oh, qué aburrido) necesita ser estable cuando sale con el par extra de electrones. De hecho, podemos predecir las características de un buen grupo de salida. Por ejemplo, el enlace al grupo lábil debe estar polarizado, y dado que el grupo de salida lleva consigo el par de electrones, el grupo debe ser estable con este par extra de electrones en él (L−). Otra forma de decir esto es que el grupo de salida debe ser electronegativo y romper elC−L vínculo debe producir una base débil. Los iones haluro son ejemplos de buenos grupos salientes, y su orden de reactividad esI−>Br−>Cl−>F−. Esta clasificación refleja sus rangos de fuerza ácida, es decir,HI es el ácido más fuerte yHF el más débil, lo que significa queF− es la base más fuerte (y por lo tanto es menos probable que se vaya)
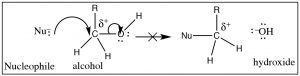
Entonces, ¿qué pasa con el oxígeno, en forma deO−H grupo alcohólico, como grupo de salida? (→) Ciertamente cumple con el requisito de que elC−O enlace sea polarizado, pero si sigues la reacción a través de ella significaría que el grupo de salida sería un ion hidróxido (−OH), una base muy fuerte. Por lo tanto, no es probable que los alcoholes (ROH) sean atacados por un nucleófilo.
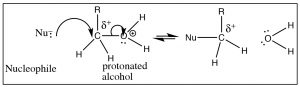
Sin embargo, hay formas de hacer que un alcohol sea reactivo. Por ejemplo, si podemos llevar a cabo la reacción en una solución ácida, el alcohol se protonará (al menos una parte del tiempo), y por lo tanto el grupo de salida será una molécula de agua, una entidad estable (→).
¿Qué hace que un buen nucleófilo?
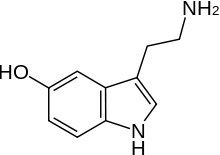
Como hemos señalado, una base de Lewis también es un nucleófilo, por lo que las tendencias que has aprendido sobre las fortalezas de las bases de Lewis también tienen para los nucleófilos. Entonces, por ejemplo, la nucleofilia disminuye a lo largo de una fila en la tabla periódica así queNH3>H2O>HF de la misma manera que lo hace la fuerza base (recuerde que esto se debe a que el par solitario está más disponible en elN que enF). Pero como esto es química orgánica, deberíamos tener algunos grupos orgánicos colgados de los nucleófilos. Entonces, por ejemplo, en lugar de un nucleófilo hidróxido, podríamos usar un nucleófilo alcóxido (por ejemplo, etóxido deCH3CH2O−Na+ sodio), o podríamos usar nucleófilos amínicos como la serotonina (el nitrógeno en elNH2 grupo aquí es más nucleófilo que elOH grupo, y elN en el anillo). Además, si comparamos nucleófilos con el mismo átomo nucleofílico, una especie cargada negativamente es más nucleofílica que la forma no cargada, entoncesOH−>H2O, yNH2−>NH3 (y por analogía cualquier derivado orgánico se comporta de la misma manera).
Además de los nucleófilos que son fácilmente reconocibles por ser bases, hay otra clase de nucleófilos que son algo diferentes; tienen un par solitario de electrones, pero no son particularmente básicos. Los ejemplos más comunes son los iones haluro, que son bases débiles y buenos grupos salientes. Entonces, surge la pregunta: ¿por qué los iones haluro son tan buenos nucleófilos? La razón de esto tiene que ver con su polarizabilidad (es decir, la medida en que una nube de electrones puede distorsionarse) del nucleófilo. Un yoduro tipo anión muy grande tiene una nube de electrones muy polarizable porque los electrones se extienden mucho más lejos del núcleo que, por ejemplo, la nube de electrones en fluoruro. Esto significa que la nube de electrones para yoduro puede comenzar la formación de enlaces parciales al carbono mucho antes que la del fluoruro, y por lo tanto el yoduro reacciona mucho más rápido que el fluoruro. [18] Esta lógica nos permite explicar por qué la nucleofilia de los iones haluro aumenta a medida que baja un grupo:I−>Br−>Cl−>F−.
Si bien volveremos a esta reacción con mayor detalle más adelante, echemos un vistazo a la gama de posibles reacciones que este esquema genérico nos permite predecir, con la salvedad de que estamos considerando sustratos de carbono simples. Reacciones como esta se denominan sustituciones nucleofílicas, porque la especie que ataca al carbono es un nucleófilo, y el efecto general de la reacción es que hemos sustituido al nucleófilo por el grupo lábil. Este ejemplo particular se llamaSN2 reacción que significa S ubstitution, N ucleophilic, 2nd Order, y volveremos a discutir la reacción con mucho más detalle más adelante.
Otro tipo de nucleófilo de carbono
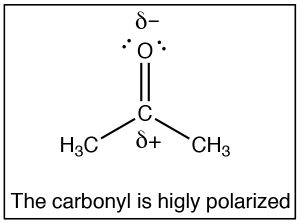
LaSN2 reacción es un pilar de la química orgánica, al variar el sustrato (electrófilo de carbono) el grupo lábil, y el nucleófilo podemos construir una gran variedad de compuestos diferentes. Otro tipo muy importante de compuesto que tiene un carbono electrófilo (es decir, un carbono que está sujeto a ataque nucleofílico) es uno que contiene un grupo carbonilo (C=O). El grupo carbonilo está altamente polarizado, con un granδ+ en el carbono. Esto puede ser racionalizado por la idea de que hay dos enlaces al oxígeno electronegativo y por lo tanto el oxígeno tiene aún más tendencia a alejar electrones del carbono que lo haría un solo oxígeno unido. Una forma de visualizar esto es dibujar estructuras de resonancia para el grupo carbonilo como se muestra, donde los electrones del doble enlace ahora se encuentran en elO. Volveremos a cómo dibujar formas de resonancia con mucho más detalle más adelante.
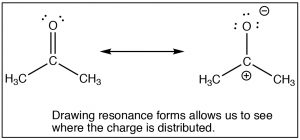
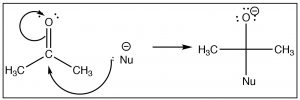
Una vez que entendemos cómo se polarizan los compuestos con grupos carbonilo, podemos predecir (al menos para el primer paso) cómo reaccionarán estos compuestos. Por ejemplo, si tenemos un nucleófilo razonablemente bueno (aquí se muestra comoNu−) podríamos predecir que atacaría al carbono carbonilo. La diferencia en esta reacción y unaSN2 reacción es que no hay grupo de salida. En cambio, los electrones de uno de losC−O enlaces se mueven hacia el oxígeno como se muestra.
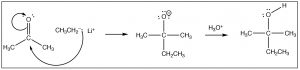
Hay varias formas en que esta reacción puede continuar, la más obvia de las cuales es que si la reacción está en contacto con un disolvente que tiene protones ácidos (por ejemplo, agua o un alcohol), elO− puede simplemente protonar en una reacción ácido-base. Como veremos más adelante, el curso de la reacción también depende de lo que sea el nucleófilo. Aquí daremos el ejemplo más simple que es la reacción de una cetona (acetona) con un nucleófilo de carbono (CH3CH2Li, etil litio). Por ahora, no nos preocuparemos por cómo hacer etil litio, ¡pero ten la seguridad de que es posible! Cuando el electrófilo de carbono cargado negativamente se agrega al carbonilo, hacemos un nuevo enlace carbono-carbono. A esto le sigue la adición de agua para protonar el oxígeno, para producir un alcohol. La reacción global es una adición nucleofílica.
Preguntas para responder
Intente predecir los resultados de estas reacciones dibujando mecanismos de empuje de flechas.
- CH3CH2I+NaOH→
- CH3Br+NaN3→
- CH3CH2Cl+NH2CH3→
- CH3OH+H+→
¿Qué nucleófilo y electrófilo reaccionarían juntos para formar estos productos?
- CH3OH+Br−
- CH3CH3NH+3+Cl−
- Construir un modelo generalizable para laSN2 reacción y explicar el papel del sustrato (el electrófilo de carbono), el grupo lábil y el nucleófilo.
Construir un modelo generalizable para la reacción de adición nucleofílica y explicar el papel del sustrato (el electrófilo de carbono) y el nucleófilo. ¿Qué grupos funcionales sufrirían una adición nucleofílica? - ¿Qué haría de un carbono en un compuesto un nucleófilo? ¿Cómo podrías hacer un carbono nucleofílico en particular?