
12.3: Optimización de las Separaciones Cromatográficas
- Page ID
- 75670

Ahora que hemos definido el factor de retención de solutos, la selectividad y la eficiencia de la columna, podemos considerar cómo afectan la resolución de dos picos de elución cercana. Debido a que los dos picos tienen tiempos de retención similares, es razonable suponer que sus anchos de pico son casi idénticos. Si el número de placas teóricas es el mismo para todos los solutos—no estrictamente cierto, pero no una mala suposición— entonces a partir de la ecuación 12.2.15, la relación t r/w es una constante. Si dos solutos tienen tiempos de retención similares, entonces sus anchos de pico deben ser similares. La ecuación 12.2.1, por lo tanto, se convierte
donde B es la elución posterior de los dos solutos. Resolver la ecuación 12.2.15 para w B y sustituirla en Ecuación\ ref {12.1} nos deja con el siguiente resultado.
\[R_{A B}=\frac{\sqrt{N_{B}}}{4} \times \frac{t_{r, B}-t_{r, A}}{t_{r, B}} \label{12.2}\]
Reorganizar la ecuación 12.2.8 nos proporciona las siguientes ecuaciones para los tiempos de retención de los solutos A y B.
\[t_{r, A}=k_{A} t_{\mathrm{m}}+t_{\mathrm{m}} \quad \text { and } \quad t_{\mathrm{r}, B}=k_{B} t_{\mathrm{m}}+t_{\mathrm{m}} \nonumber\]
Después de sustituir estas ecuaciones en la Ecuación\ ref {12.2} y simplificarlas, tenemos
\[R_{A B}=\frac{\sqrt{N_{B}}}{4} \times \frac{k_{B}-k_{A}}{1+k_{B}} \nonumber\]
Finalmente, podemos eliminar el factor de retención del soluto A sustituyendo en la ecuación 12.2.9. Después de reorganizar, terminamos con la siguiente ecuación para la resolución entre los picos cromatográficos para los solutos A y B.
Además de la resolución, otro factor importante en la cromatografía es la cantidad de tiempo necesario para eluir un par de solutos, que podemos aproximar utilizando el tiempo de retención para el soluto B.
donde u es la velocidad de la fase móvil.
Aunque la Ecuación\ ref {12.3} es útil para considerar cómo un cambio en N,\(\alpha\), o k afecta cualitativamente la resolución, lo que se adapta a nuestro propósito aquí, es menos útil para hacer predicciones cuantitativas precisas de resolución, particularmente para valores más pequeños de N y para valores mayores de R. Para predicciones más precisas usa la ecuación
\[R_{A B}=\frac{\sqrt{N}}{4} \times(\alpha-1) \times \frac{k_{B}}{1+k_{\mathrm{avg}}} \nonumber\]
donde k avg es (k A + k B) /2. Para una derivación de esta ecuación y para una discusión más profunda de la resolución en cromatografía en columna, véase Foley, J. P. “Resolution Equings for Column Chromatography”, Analyst, 1991, 116, 1275-1279.
La ecuación\ ref {12.3} y la ecuación\ ref {12.4} contienen términos que corresponden a la eficiencia de la columna, la selectividad y el factor de retención del soluto. Podemos variar estos términos, de manera más o menos independiente, para mejorar la resolución y el tiempo de análisis. El primer término, que es función del número de placas teóricas (para la Ecuación\ ref {12.3}) o la altura de una placa teórica (para la Ecuación\ ref {12.4}), explica el efecto de la eficiencia de la columna. El segundo término es una función\(\alpha\) y explica la influencia de la selectividad de la columna. Finalmente, el tercer término en ambas ecuaciones es una función de k B y explica el efecto del factor de retención del soluto B. Una discusión sobre cómo podemos usar estos parámetros para mejorar la resolución es el tema del resto de esta sección.
Uso del factor de retención para optimizar la resolución
Una de las formas más sencillas de mejorar la resolución es ajustar el factor de retención para el soluto B. Si todos los demás términos de la Ecuación\ ref {12.3} permanecen constantes, un incremento en k B mejorará la resolución. Como lo muestra la curva verde en la Figura 12.3.1 , sin embargo, la mejora es mayor si el valor inicial de k B es pequeño. Una vez que k B supera un valor de aproximadamente 10, un aumento adicional produce solo una mejora marginal en la resolución. Por ejemplo, si el valor original de k B es 1, aumentar su valor a 10 da una mejora de 82% en la resolución; otro incremento a 15 proporciona una mejora neta en la resolución de sólo 87.5%.
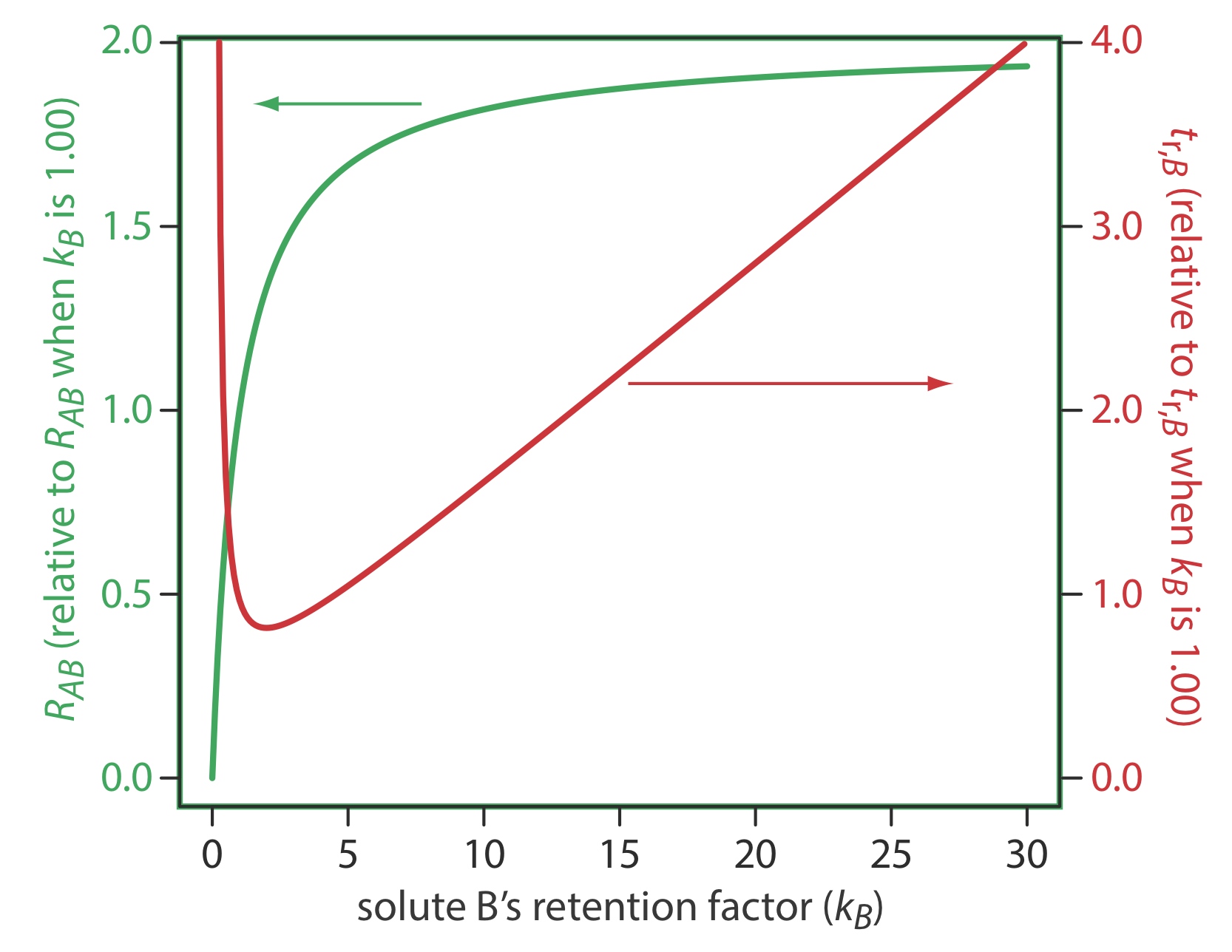
Cualquier mejora en la resolución por aumentar el valor de k B generalmente viene a costa de un tiempo de análisis más largo. La curva roja en la Figura 12.3.1 muestra el cambio relativo en el tiempo de retención para el soluto B en función de su factor de retención. Tenga en cuenta que el tiempo mínimo de retención es para k B = 2. Aumentar k B de 2 a 10, por ejemplo, aproximadamente duplica el tiempo de retención del soluto B.
La relación entre el factor de retención y el tiempo de análisis en la Figura {{template.index (ID:1)} funciona a nuestro favor si una separación produce una resolución aceptable con un k B grande. En este caso podemos disminuir k B con poca pérdida de resolución y con un tiempo de análisis significativamente más corto.
Para aumentar k B sin cambiar la selectividad\(\alpha\), cualquier cambio en las condiciones cromatográficas debe dar como resultado un aumento general no selectivo en el factor de retención para ambos solutos. En cromatografía de gases, podemos lograr esto disminuyendo la temperatura de la columna. Debido a que la presión de vapor de un soluto es menor a temperaturas más bajas, pasa más tiempo en la fase estacionaria y tarda más en eluirse. En la cromatografía líquida, la forma más fácil de aumentar el factor de retención de un soluto es usar una fase móvil que sea un disolvente más débil. Cuando la fase móvil tiene una menor fuerza de solvente, los solutos pasan proporcionalmente más tiempo en la fase estacionaria y tardan más en eluirse.
Ajustar el factor de retención para mejorar la resolución entre un par de solutos puede conducir a tiempos de retención inaceptablemente largos para otros solutos. Por ejemplo, supongamos que necesitamos analizar una mezcla de cuatro componentes con resolución basal y con un tiempo de ejecución inferior a 20 min. Nuestra elección inicial de condiciones da el cromatograma en la Figura 12.3.2 a. Aunque separamos con éxito los componentes 3 y 4 en 15 min, fallamos en separar los componentes 1 y 2. Ajustar las condiciones para mejorar la resolución de los dos primeros componentes al aumentar k 2 proporciona una buena separación de los cuatro componentes, pero el tiempo de ejecución es demasiado largo (Figura 12.3.2 b). Este problema de encontrar un solo conjunto de condiciones de operación aceptables se conoce como el problema general de elución.
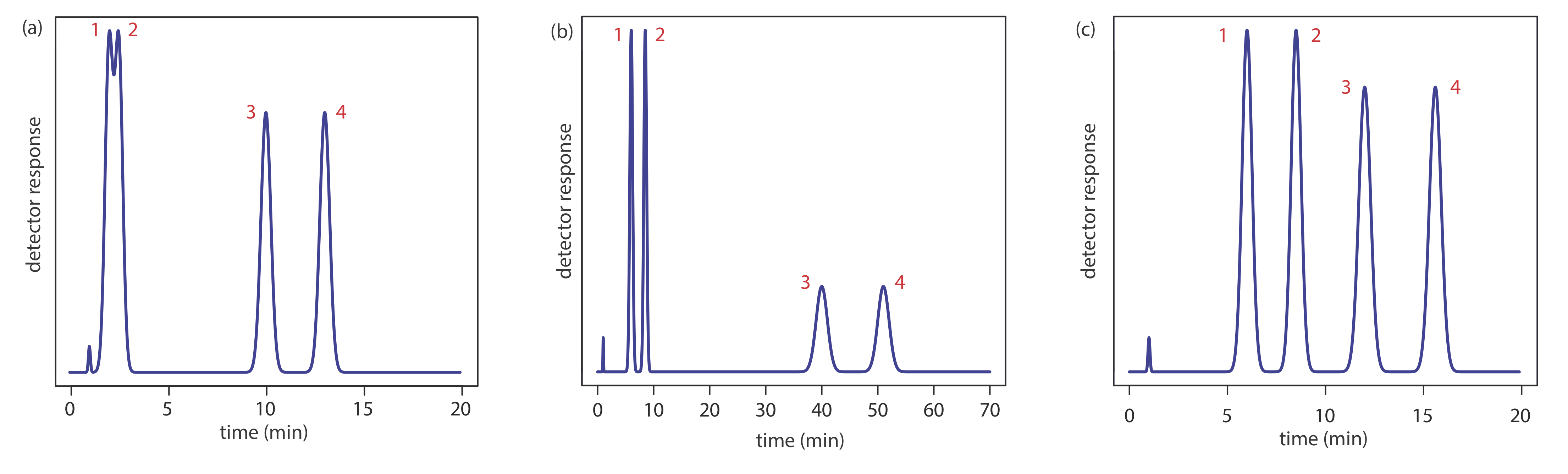
Una solución al problema general de elución es hacer ajustes incrementales al factor de retención a medida que se produce la separación. Al inicio de la separación establecemos las condiciones cromatográficas iniciales para optimizar la resolución de solutos de elución temprana. A medida que avanza la separación, ajustamos las condiciones cromatográficas para disminuir el factor de retención y, por tanto, disminuir el tiempo de retención, para cada uno de los solutos que eluyen posteriormente (Figura 12.3.2 c). En cromatografía de gases esto se logra mediante programación de temperatura. La temperatura inicial de la columna se selecciona de tal manera que los primeros solutos a eluir se resuelven completamente. Luego se aumenta la temperatura, ya sea de forma continua o por etapas, para desprender los componentes que eluyen posteriormente con una resolución aceptable y un tiempo de análisis razonable. En cromatografía líquida se obtiene el mismo efecto aumentando la fuerza de elución del disolvente. Esto se conoce como elución en gradiente. Tendremos más que decir sobre cada uno de estos en secciones posteriores de este capítulo.
Uso de la selectividad para optimizar la resolución
Un segundo enfoque para mejorar la resolución es ajustar la selectividad,\(\alpha\). De hecho, por\(\alpha \approx 1\) lo general no es posible mejorar la resolución ajustando el factor de retención de soluto, k B, o la eficiencia de la columna, N. Un cambio en\(\alpha\) muchas veces tiene un efecto más dramático en la resolución que un cambio en k B. Por ejemplo, cambiar\(\alpha\) de 1.1 a 1.5, mientras se mantienen constantes todos los demás términos, mejora la resolución en un 267%. En cromatografía de gases, ajustamos\(\alpha\) cambiando la fase estacionaria; en cromatografía líquida, cambiamos la composición de la fase móvil para ajustarla\(\alpha\).
Para cambiar\(\alpha\) necesitamos ajustar selectivamente los factores individuales de retención de solutos. La Figura 12.3.3 muestra una posible aproximación para la separación por cromatografía líquida de una mezcla de ácidos benzoicos sustituidos. Debido a que el tiempo de retención de la forma ácida débil de un compuesto y su forma de base débil son diferentes, su tiempo de retención variará con el pH de la fase móvil, como se muestra en la Figura 12.3.3 a. las intersecciones de las curvas en la Figura {{template.index (ID:3)} a muestran valores de pH donde dos solutos eluyen conjuntamente. Por ejemplo, a un pH de 3.8, el ácido tereftálico y el ácido p-hidroxibenzoico eluyen como un único pico cromatográfico.
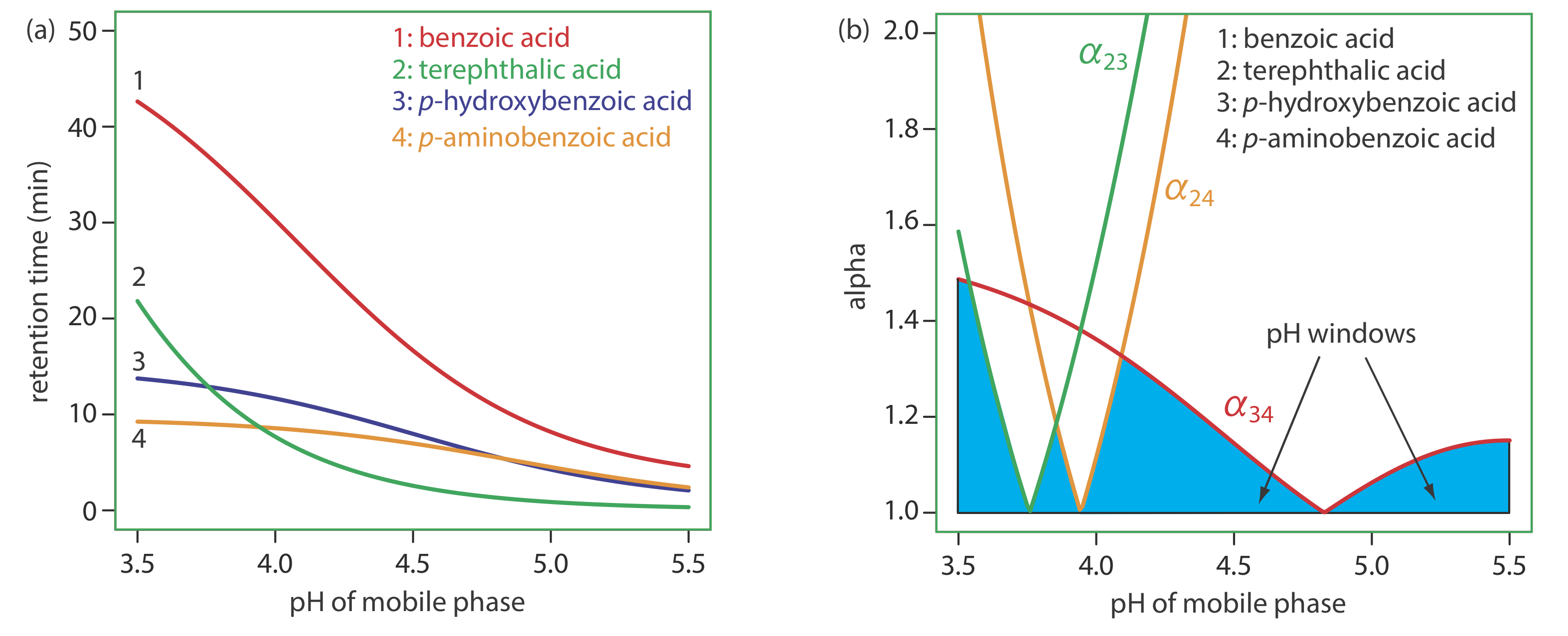
La figura 12.3.3 a muestra que hay muchos valores de pH donde es posible cierta separación. Para encontrar la separación óptima, trazamos una para cada par de solutos. Las curvas roja, verde y naranja en la Figura 12.3.3 b muestran la variación en a con pH para los tres pares de solutos que son más difíciles de separar (para todos los demás pares de solutos,\(\alpha\) > 2 en todos los niveles de pH). El sombreado azul muestra ventanas de valores de pH en las que al menos es posible una separación parcial, esta cifra a veces se denomina diagrama de ventanas, y el punto más alto en cada ventana da el pH óptimo dentro de ese rango. La mejor separación general es el punto más alto en cualquier ventana, que, para este ejemplo, es un pH de 3.5. Debido a que el tiempo de análisis a este pH es superior a 40 min (Figura 12.3.3 a), elegir un pH entre 4.1—4.4 podría producir una separación aceptable con un tiempo de análisis mucho más corto.
Usemos ácido benzoico, C 6 H 5 COOH, para explicar por qué el pH puede afectar el tiempo de retención de un soluto. La separación utiliza una fase móvil acuosa y una fase estacionaria no polar. A pH más bajos, el ácido benzoico se encuentra predominantemente en su forma de ácido débil, C 6 H 5 COOH, y se divide fácilmente en la fase estacionaria no polar. A pH más básicos, sin embargo, el ácido benzoico se encuentra en su forma de base débil, C 6 H 5 COO —. Debido a que ahora lleva una carga, su solubilidad en la fase móvil aumenta y su solubilidad en la fase estacionaria no polar disminuye. Como resultado, pasa más tiempo en la fase móvil y tiene un tiempo de retención más corto.
Si bien la forma habitual de ajustar el pH es cambiar la concentración de agentes amortiguadores, también es posible ajustar el pH cambiando la temperatura de la columna porque el valor p K a de un soluto es dependiente del pH; para una revisión, ver Gagliardi, L. G.; Tascon, M.; Castells, C. B. “Efecto de Temperatura en Equilibrios Ácido-Base en Técnicas de Separación: Una Revisión,” Anal. Chim. Acta, 2015, 889, 35—57.
Uso de la eficiencia de columna para optimizar la resolución
Un tercer enfoque para mejorar la resolución es ajustar la eficiencia de la columna aumentando el número de placas teóricas, N. Si tenemos valores para k B y\(\alpha\), entonces podemos usar la Ecuación\ ref {12.3} para calcular el número de placas teóricas para cualquier resolución. Table 12.3.1 proporciona algunos valores representativos. Por ejemplo, si\(\alpha\) = 1.05 y k B = 2.0, una resolución de 1.25 requiere aproximadamente 24 800 placas teóricas. Si nuestra columna proporciona solo 12 400 placas, la mitad de lo que se necesita, entonces no es posible una separación. ¿Cómo podemos duplicar el número de planchas teóricas? La forma más fácil es duplicar la longitud de la columna, aunque esto también duplica el tiempo de análisis. Un mejor enfoque es cortar la altura de una placa teórica, H, a la mitad, proporcionando la resolución deseada sin cambiar el tiempo de análisis. Aún mejor, si podemos disminuir H en más de 50%, puede ser posible lograr la resolución deseada con un tiempo de análisis aún más corto al disminuir también k B o\(\alpha\).
Para disminuir la altura de una placa teórica necesitamos entender los factores experimentales que afectan el ensanchamiento de banda. Existen varios tratamientos teóricos de ampliación de banda. Consideraremos un enfoque que considera cuatro contribuciones: variaciones en la longitud de trayectoria, difusión longitudinal, transferencia de masa en la fase estacionaria y transferencia de masa en la fase móvil.
Varias trayectorias: variaciones en la longitud de la trayectoria
A medida que las moléculas de soluto pasan por la columna recorren caminos que difieren en longitud. Debido a esta diferencia en la longitud de la ruta, dos moléculas de soluto que ingresan a la columna al mismo tiempo saldrán de la columna en diferentes momentos. El resultado, como se muestra en la Figura 12.3.4 , es una ampliación del perfil del soluto en la columna. La contribución de múltiples caminos a la altura de una placa teórica, H p, es
\[H_{p}=2 \lambda d_{p} \label{12.5}\]
donde d p es el diámetro promedio del material particulado de empaque y\(\lambda\) es una constante que da cuenta de la consistencia del empaque. Un rango más pequeño de tamaños de partícula y un empaque más consistente producen un valor más pequeño para\(\lambda\). Para una columna sin material de empaque, H p es cero y no hay contribución al ensanchamiento de banda desde múltiples trayectorias.
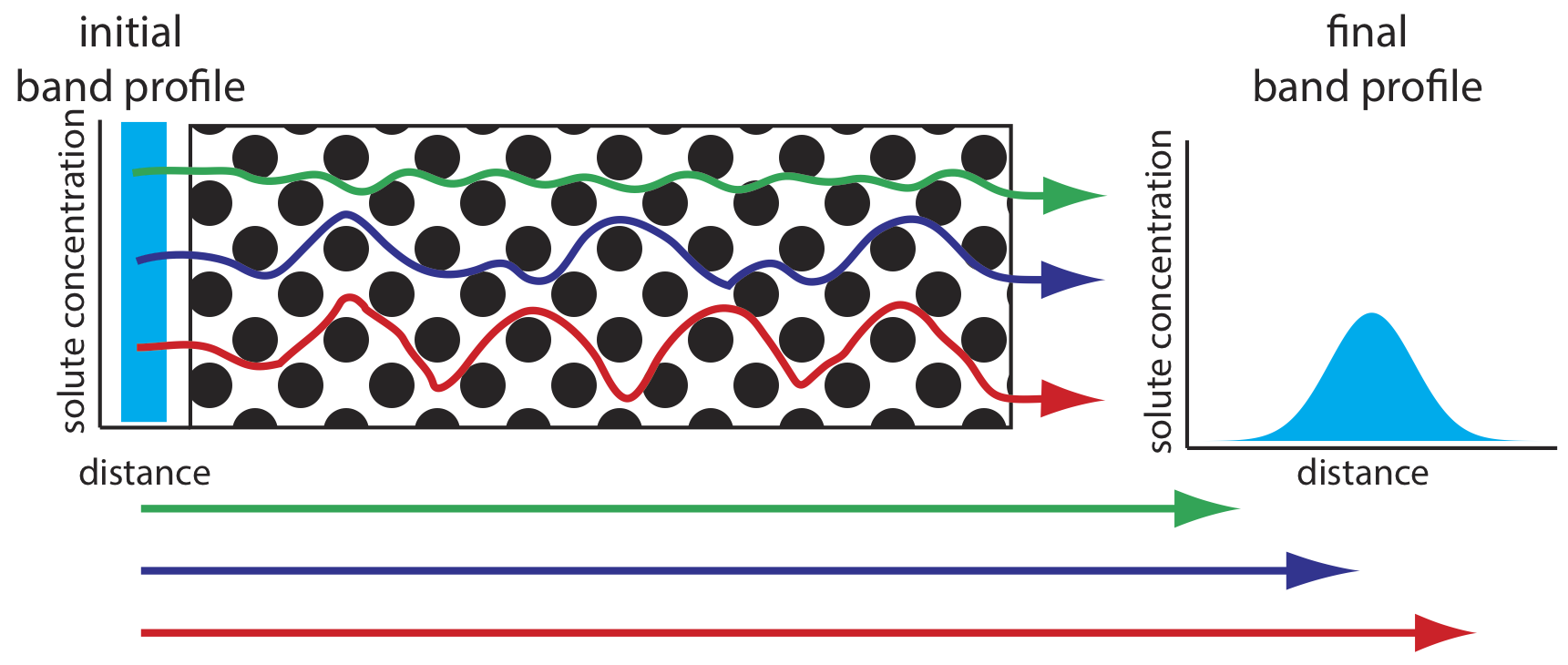
Un empaquetamiento inconsistente crea canales que permiten que algunas moléculas de soluto viajen rápidamente a través de la columna. También puede crear bolsas que atrapan temporalmente algunas moléculas de soluto, ralentizando su progreso a través de la columna. Un empaque más uniforme minimiza estos problemas.
Difusión longitudinal
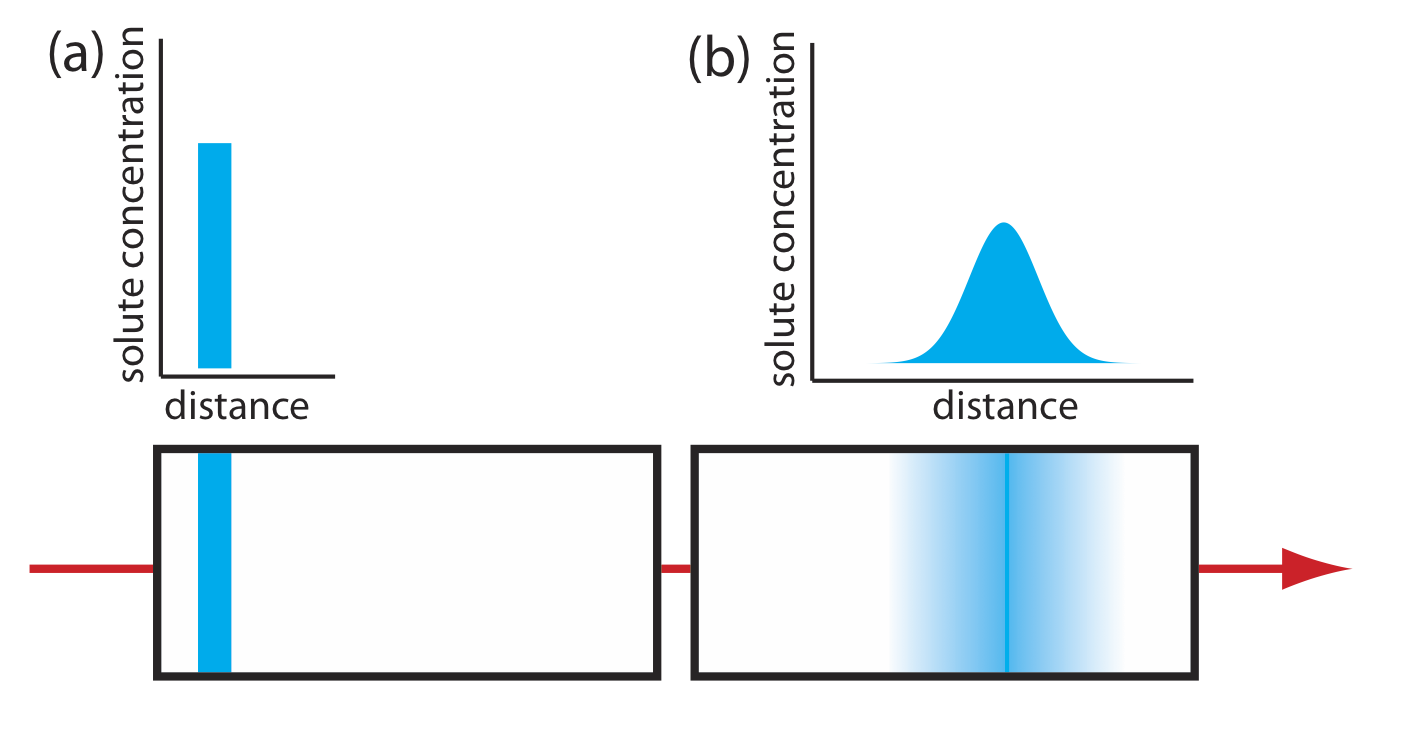
La segunda contribución al ensanchamiento de banda es el resultado de la difusión longitudinal del soluto en la fase móvil. Las moléculas de soluto están en constante movimiento, difundiéndose desde regiones de mayor concentración de soluto a regiones donde la concentración de soluto es menor. El resultado es un aumento en el ancho de banda del soluto (Figura 12.3.5 ). La contribución de la difusión longitudinal a la altura de una placa teórica, H d, es
\[H_{d}=\frac{2 \gamma D_{m}}{u} \label{12.6}\]
donde D m es el coeficiente de difusión del soluto en la fase móvil, u es la velocidad de la fase móvil y\(\gamma\) es una constante relacionada con la eficiencia del empaquetamiento de la columna. Obsérvese que el efecto de H d sobre el ensanchamiento de banda es inversamente proporcional a la velocidad de la fase móvil: una mayor velocidad proporciona menos tiempo para la difusión longitudinal. Debido a que el coeficiente de difusión de un soluto es mayor en la fase gaseosa que en una fase líquida, la difusión longitudinal es un problema más grave en la cromatografía de gases.
Transferencia Masiva
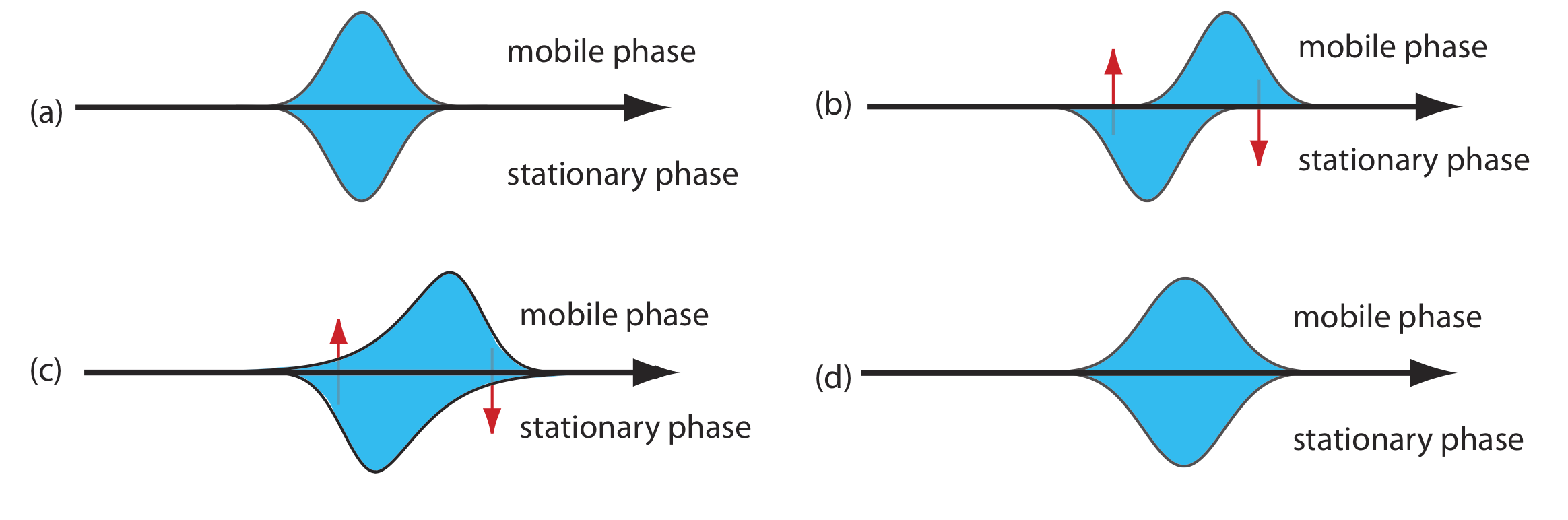
A medida que el soluto pasa por la columna se mueve entre la fase móvil y la fase estacionaria. A este movimiento lo llamamos transferencia de masa entre fases. Como se muestra en la Figura 12.3.6 , el ensanchamiento de banda ocurre si el movimiento del soluto dentro de la fase móvil o dentro de la fase estacionaria no es lo suficientemente rápido como para mantener un equilibrio en su concentración entre las dos fases. En promedio, una molécula de soluto en la fase móvil baja por la columna antes de pasar a la fase estacionaria. Una molécula de soluto en la fase estacionaria, por otro lado, tarda más de lo esperado en volver a la fase móvil. Las contribuciones de transferencia de masa en la fase estacionaria, H s, y transferencia de masa en la fase móvil, H m, están dadas por las siguientes ecuaciones
\[H_{s}=\frac{q k d_{f}^{2}}{(1+k)^{2} D_{s}} u \label{12.7}\]
\[H_{m}=\frac{f n\left(d_{p}^{2}, d_{c}^{2}\right)}{D_{m}} u \label{12.8}\]
donde d f es el espesor de la fase estacionaria, d c es el diámetro de la columna, D s y D m son los coeficientes de difusión para el soluto en la fase estacionaria y la fase móvil, k es el factor de retención del soluto y q es una constante relacionada con el material de relleno de la columna. Aunque no se conoce la forma exacta de H m, es una función del tamaño de partícula y el diámetro de la columna. Tenga en cuenta que el efecto de H s y H m sobre el ensanchamiento de banda es directamente proporcional a la velocidad de la fase móvil porque una velocidad menor proporciona más tiempo para la transferencia de masa.
La abreviatura fn en Ecuación\ ref {12.7} significa “es una función de”.
Poniéndolo todo junto
La altura de una placa teórica es una suma de las contribuciones de cada uno de los términos que afectan el ensanchamiento de banda.
\[H=H_{p}+H_{d}+H_{s}+H_{m} \label{12.9}\]
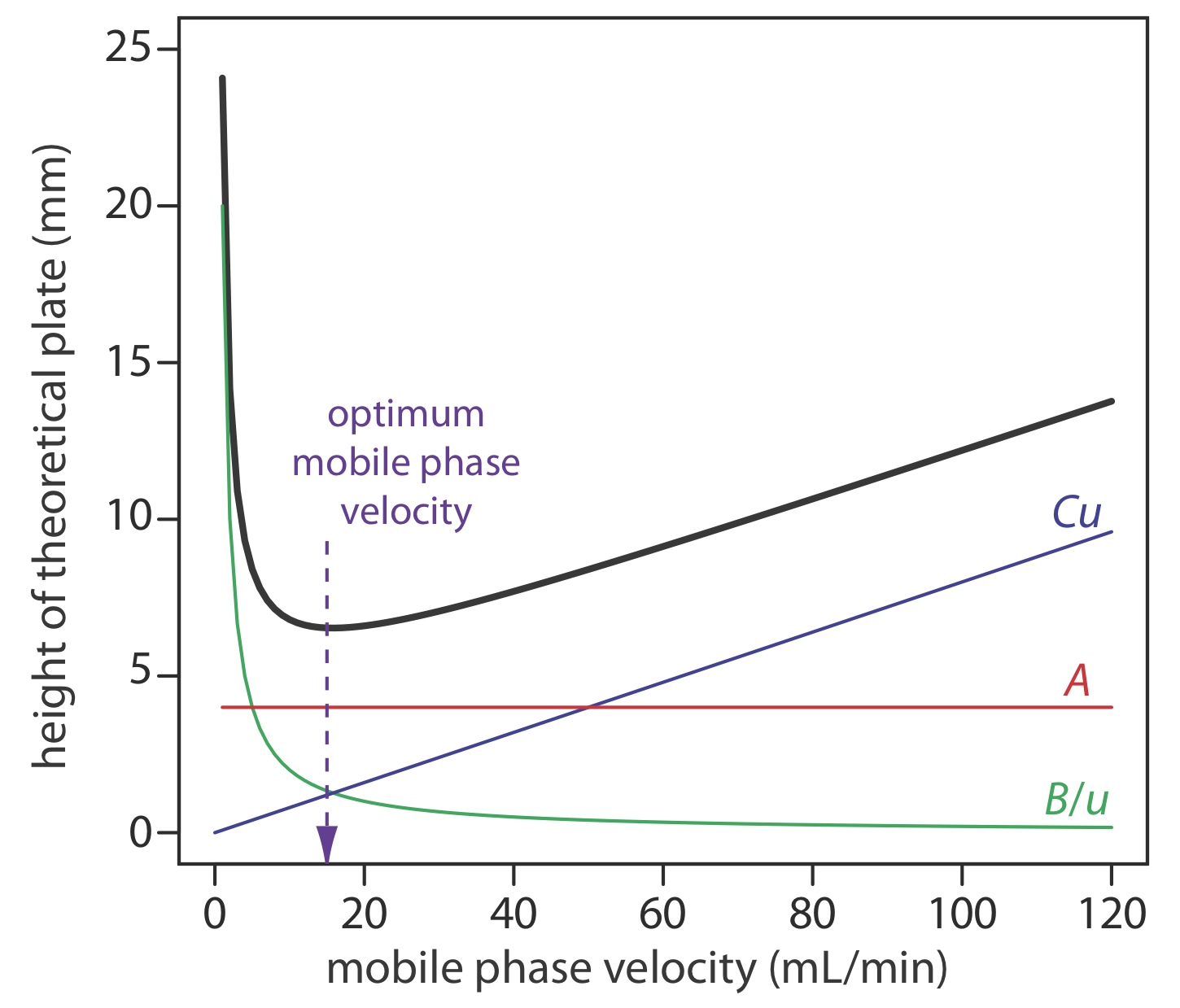
Una forma alternativa de esta ecuación es la ecuación de van Deemter
\[H=A+\frac{B}{u}+C u \label{12.10}\]
que enfatiza la importancia de la velocidad de la fase móvil. En la ecuación de van Deemter, A da cuenta de la contribución de múltiples caminos (H p), B/u representa la contribución de la difusión longitudinal (H d), y Cu representa la contribución combinada de transferencia de masa en la fase estacionaria y en la fase móvil (H s y H m).
Existe cierto desacuerdo sobre la mejor ecuación para describir la relación entre la altura de la placa y la velocidad de la fase móvil [Hawkes, S. J. J. Chem. Educ. 1983, 60, 393—398]. Además de la ecuación de van Deemter, otras ecuaciones incluyen
\[H=\frac{B}{u}+\left(C_s+C_{m}\right) u \nonumber\]
donde C s y C m son los términos de transferencia de masa para la fase estacionaria y la fase móvil y
\[H=A u^{1 / 3}+\frac{B}{u}+C u \nonumber\]
Las tres ecuaciones, y otras, han sido utilizadas para caracterizar sistemas cromatográficos, sin que una sola ecuación proporcione la mejor explicación en cada caso [Kennedy, R. T.; Jorgenson, J. W. Anal. Chem. 1989, 61, 1128—1135].
Para aumentar el número de placas teóricas sin aumentar la longitud de la columna, necesitamos disminuir uno o más de los términos en la Ecuación\ ref {12.9}. La forma más fácil de disminuir H es ajustar la velocidad de la fase móvil. Para velocidades de fase móvil más pequeñas, la eficiencia de la columna está limitada por la difusión longitudinal, y para velocidades de fase móviles más altas la eficiencia está limitada por los dos términos de transferencia de masa. Como se muestra en la Figura 12.3.7 —que utiliza la ecuación van Deemter— la velocidad óptima de fase móvil es la mínima en una gráfica de H en función de u.
Los parámetros restantes que afectan a los términos de la Ecuación\ ref {12.9} son funciones de las propiedades de la columna y sugieren otros posibles enfoques para mejorar la eficiencia de la columna. Por ejemplo, tanto H p como H m son una función del tamaño de las partículas utilizadas para empaquetar la columna. La disminución del tamaño de partícula, por lo tanto, es otro método útil para mejorar la eficiencia.
Para una discusión más detallada de las formas de evaluar la calidad de una columna, véase Desmet, G.; Caooter, D.; Broeckhaven, K. “Métodos de representación de datos gráficos para evaluar la calidad de columnas LC”, Anal. Chem. 2015, 87, 8593—8602.
Quizás el avance más importante en las columnas de cromatografía es el desarrollo de columnas tubulares abiertas o capilares. Estas columnas tienen diámetros muy pequeños (d c ≈ 50—500 μm) y no contienen material de empaque (d p = 0). En cambio, la pared interior de la columna capilar está recubierta con una película delgada de la fase estacionaria. La altura de placa se reduce porque la contribución a H de H p (Ecuación\ ref {12.5}) desaparece y la contribución de H m (Ecuación\ ref {12.8}) se vuelve más pequeña. Debido a que la columna no contiene ningún material de relleno sólido, se necesita menos presión para mover la fase móvil a través de la columna, lo que permite columnas más largas. La combinación de una columna más larga y una altura menor para una placa teórica aumenta aproximadamente el número de placas teóricas\(100 \times\). Las columnas capilares no están exentas de desventajas. Debido a que son mucho más estrechas que las columnas empaquetadas, requieren una cantidad significativamente menor de muestra, lo que puede ser difícil de inyectar de manera reproducible. Otro enfoque para mejorar la resolución es utilizar películas delgadas de fase estacionaria, lo que disminuye la contribución a H de H s (Ecuación\ ref {12.7}).
Cuanto más pequeñas son las partículas, más presión se necesita para empujar la fase móvil a través de la columna. Como resultado, para cualquier forma de cromatografía existe un límite práctico para el tamaño de partícula.