
12.4: Cromatografía de gases
- Page ID
- 75654

En cromatografía de gases (GC) inyectamos la muestra, que puede ser un gas o un líquido, en una fase móvil gaseosa (a menudo llamada gas portador). La fase móvil transporta la muestra a través de una columna empaquetada o capilar que separa los componentes de la muestra en función de su capacidad de partición entre la fase móvil y la fase estacionaria. La Figura 12.4.1 muestra un ejemplo de un cromatógrafo de gases típico, que consta de varios componentes clave: un suministro de gas comprimido para la fase móvil; un inyector calentado, que volatiliza rápidamente los componentes en una muestra líquida; una columna, que se coloca dentro de un horno cuya temperatura nosotros puede controlar durante la separación; y un detector para monitorear el eluyente a medida que sale de la columna. Consideremos cada uno de estos componentes.

Fase Móvil
Las fases móviles más comunes para la cromatografía de gases son He, Ar y N 2, las cuales tienen la ventaja de ser químicamente inertes tanto hacia la muestra como hacia la fase estacionaria. La elección del gas portador a menudo está determinada por las necesidades del detector del instrumento. Para una columna empaquetada, la velocidad de la fase móvil suele ser de 25—150 ml/min. El caudal típico para una columna capilar es de 1—25 ml/min.
Columnas Cromatográficas
Hay dos clases amplias de columnas cromatográficas: columnas empaquetadas y columnas capilares. En general, una columna empaquetada puede manejar muestras más grandes y una columna capilar puede separar mezclas más complejas.
Columnas empaquetadas
Las columnas empaquetadas están construidas de vidrio, acero inoxidable, cobre o aluminio, y típicamente tienen una longitud de 2 a 6 m con diámetros internos de 2 a 4 mm. La columna se rellena con un soporte sólido particulado, con diámetros de partícula que van desde 37—44 μm hasta 250—354 μm. La figura 12.4.2 muestra un ejemplo típico de una columna empaquetada.

El soporte particulado más utilizado es la tierra de diatomeas, la cual está compuesta por los esqueletos de sílice de las diatomeas. Estas partículas son muy porosas, con áreas superficiales que van desde 0.5—7.5 m 2 /g, lo que proporciona un amplio contacto entre la fase móvil y la fase estacionaria. Cuando se hidroliza, la superficie de una tierra de diatomeas contiene grupos silanol (—SiOH), que sirven como sitios activos para absorber moléculas de soluto en cromatografía gas-sólido (GSC).
En cromatografía gas-líquido (GLC), recubrimos el material de empaque con una fase móvil líquida. Para evitar que el material de empaque sin recubrimiento adsorba solutos, lo que degrada la calidad de la separación, los silanoles superficiales se desactivan haciéndolos reaccionar con dimetildiclorosilano y enjuagándolos con un alcohol, típicamente metanol, antes de recubrir las partículas con fase estacionaria.
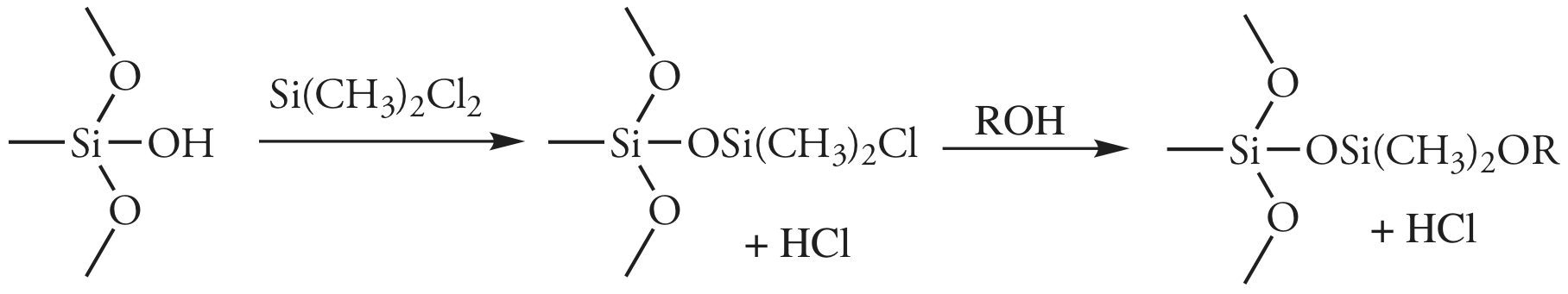
La figura 12.4.2 , por ejemplo, tiene aproximadamente 1800 placas/m, o un total de aproximadamente 3600 placas teóricas. Si asumimos un V max/V min ≈ 50, entonces tiene una capacidad pico (ecuación 12.2.16) de
\[n_{c}=1+\frac{\sqrt{3600}}{4} \ln (50) \approx 60 \nonumber\]
Columnas Capilares
Una columna capilar o tubular abierta se construye a partir de sílice fundida y se recubre con un recubrimiento polimérico protector. Las columnas varían de 15—100 m de longitud con un diámetro interno de aproximadamente 150—300 μm. La figura 12.4.3 muestra un ejemplo de una columna capilar típica.

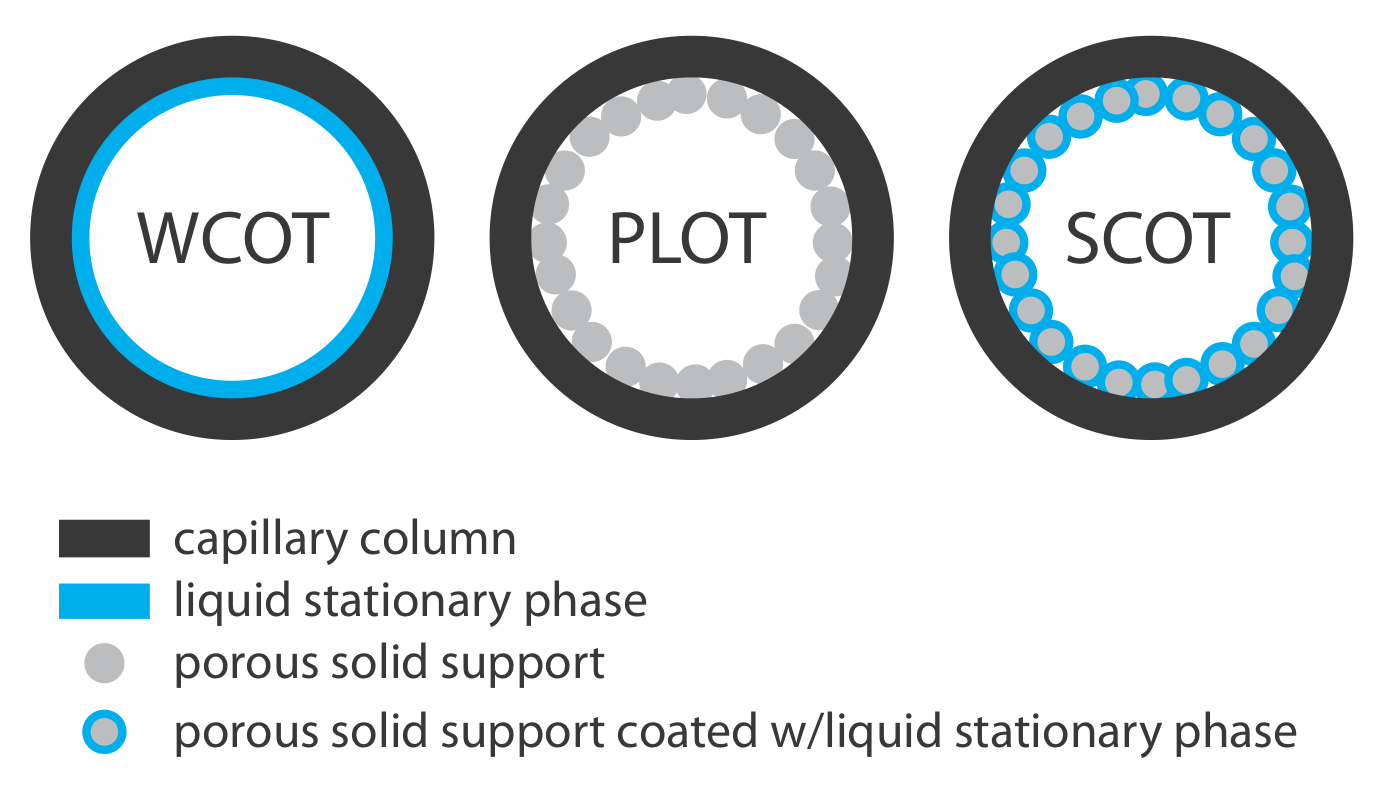
Las columnas capilares son de tres tipos principales. En una columna tubular abierta recubierta de pared (WCOT) se recubre una capa delgada de fase estacionaria, típicamente de 0.25 nm de espesor, sobre la pared interna del capilar. En una columna tubular abierta de capa porosa (PLOT), un soporte sólido poroso (alúmina, gel de sílice y tamices moleculares son ejemplos típicos) se une a la pared interna del capilar. Una columna tubular abierta recubierta con soporte (SCOT) es una columna PLOT que incluye una fase estacionaria líquida. La Figura 12.4.4 muestra las diferencias entre estos tipos de columnas capilares.
Una columna capilar proporciona una mejora significativa en la eficiencia de separación porque tiene más placas teóricas por metro y es más larga que una columna empaquetada. Por ejemplo, la columna capilar en la Figura 12.4.3 tiene casi 4300 placas/m, o un total de 129 000 placas teóricas. Si asumimos un V max/V min ≈ 50, entonces tiene una capacidad pico de aproximadamente 350. Por otro lado, una columna empaquetada puede manejar una muestra más grande. Debido a su diámetro más pequeño, una columna capilar requiere una muestra más pequeña, típicamente menos de 10 —2 μL.
Fases estacionarias para cromatografía gas-líquido
El orden de elución en la cromatografía gas-líquido depende de dos factores: el punto de ebullición de los solutos y la interacción entre los solutos y la fase estacionaria. Si los componentes de una mezcla tienen puntos de ebullición significativamente diferentes, entonces la elección de la fase estacionaria es menos crítica. Si dos solutos tienen puntos de ebullición similares, entonces una separación es posible solo si la fase estacionaria interactúa selectivamente con uno de los solutos. Como regla general, los solutos no polares se separan más fácilmente cuando se usa una fase estacionaria no polar, y los solutos polares son más fáciles de separar cuando se usa una fase estacionaria polar.
Existen varios criterios importantes para elegir una fase estacionaria: no debe reaccionar con los solutos, debe ser térmicamente estable, debe tener una volatilidad baja y debe tener una polaridad que sea apropiada para los componentes de la muestra. Table 12.4.1 resume las propiedades de varias fases estacionarias populares.
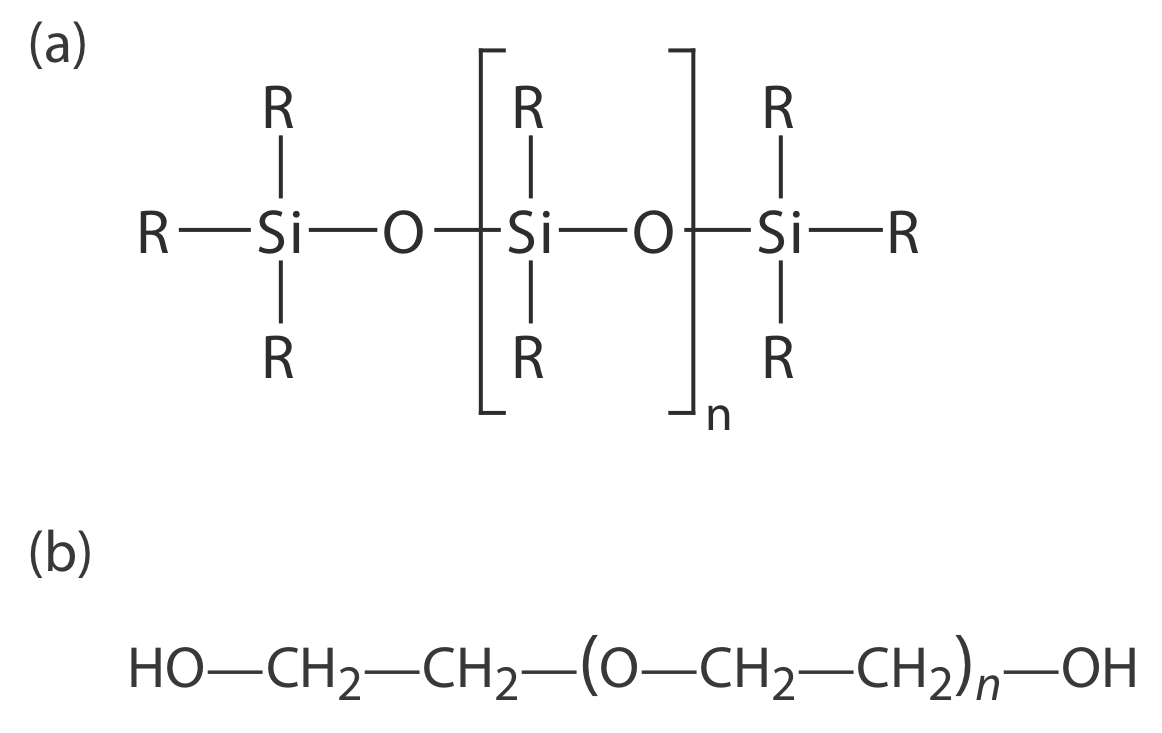
Muchas fases estacionarias tienen la estructura general mostrada en la Figura {{template.index (ID:5)} a. Una fase estacionaria de polidimetilsiloxano, en la que todos los grupos —R son grupos metilo, —CH 3, es no polar y a menudo hace una buena primera elección para una nueva separación. El orden de elución cuando se usa polidimetilsiloxano suele seguir los puntos de ebullición de los solutos, eluyendo primero los solutos de menor punto de ebullición. Reemplazar algunos de los grupos metilo con otros sustituyentes aumenta la polaridad de la fase estacionaria y proporciona mayor selectividad. Por ejemplo, reemplazar 50% de los grupos —CH 3 con grupos fenilo, —C 6 H 5, produce una fase estacionaria ligeramente polar. La polaridad creciente se proporciona sustituyendo trifluoropropilo, —C 3 H 6 CF y cianopropilo, —C 3 H 6 CN, grupos funcionales, o mediante el uso de una fase estacionaria de polietilenglicol (Figura {{template.index (ID:5)}} b).
Un problema importante con todas las fases estacionarias líquidas es su tendencia a eluir, o sangrar de la columna cuando se calienta. Los límites de temperatura en la Tabla 12.4.1 minimizan esta pérdida de fase estacionaria. Las columnas capilares con fases estacionarias unidas o reticuladas proporcionan una estabilidad superior. Una fase estacionaria unida se une químicamente a la superficie de sílice del capilar. La reticulación, que se realiza después de que la fase estacionaria está en la columna capilar, une cadenas poliméricas separadas para proporcionar una mayor estabilidad.
Otra consideración importante es el grosor de la fase estacionaria. De la ecuación 12.3.7 sabemos que la eficiencia de separación mejora con películas más delgadas de fase estacionaria. El espesor más común es de 0.25 μm, aunque una película más gruesa es útil para solutos altamente volátiles, como los gases, debido a que tiene una mayor capacidad para retener dichos solutos. Las películas más delgadas se utilizan al separar solutos de baja volatilidad, como los esteroides.
Algunas fases estacionarias aprovechan la selectividad química. Las más notables son las fases estacionarias que contienen grupos funcionales quirales, los cuales se utilizan para separar enantiómeros [Hinshaw, J. V. LC . GC 1993, 11, 644—648].
Introducción a la muestra
Tres factores determinan cómo se introduce una muestra en el cromatógrafo de gases. Primero, todos los componentes de la muestra deben ser volátiles. Segundo, los analitos deben estar presentes en una concentración apropiada. Finalmente, el proceso físico de inyección de la muestra no debe degradar la separación. Cada una de estas necesidades se considera en esta sección.
Preparación de una muestra volátil
No todas las muestras pueden inyectarse directamente en un cromatógrafo de gases. Para desplazarse por la columna, los componentes de la muestra deben ser suficientemente volátiles. Un soluto de baja volatilidad, por ejemplo, puede ser retenido por la columna y continuar eluyendo durante el análisis de muestras posteriores. Un soluto no volátil se condensará en la parte superior de la columna, degradando el rendimiento de la columna.
Podemos separar los analitos volátiles de una muestra de sus componentes no volátiles utilizando cualquiera de las técnicas de extracción descritas en el Capítulo 7. Una extracción líquido-líquido de analitos de una matriz acuosa en cloruro de metileno u otro disolvente orgánico es una elección común. Las extracciones en fase sólida también se utilizan para eliminar los componentes no volátiles de una muestra.
Un enfoque atractivo para aislar analitos es una microextracción en fase sólida (SPME). En un enfoque, que se ilustra en la Figura 12.4.6 , se coloca una fibra de sílice fundida dentro de una aguja de jeringa. La fibra, que se recubre con una película delgada de un material adsorbente, como el polidimetilsiloxano, se baja en la muestra presionando un émbolo y se expone a la muestra por un tiempo predeterminado. Después de retirar la fibra en la aguja, se transfiere al cromatógrafo de gases para su análisis.

Dos métodos adicionales para aislar analitos volátiles son un muestreo de purga y trampa y espacio de cabeza. En una purga y trampa, burbujeamos un gas inerte, como He o N 2, a través de la muestra, liberando o purgando los compuestos volátiles. Estos compuestos son transportados por el gas de purga a través de una trampa que contiene un material absorbente, como Tenax, donde son retenidos. Calentar la trampa y retrolavado con gas portador transfiere los compuestos volátiles al cromatógrafo de gases. En el muestreo del espacio de cabeza colocamos la muestra en un vial cerrado con un espacio aéreo superpuesto. Después de dejar tiempo para que los analitos volátiles se equilibren entre la muestra y el aire suprayacente, utilizamos una jeringa para extraer una porción de la fase de vapor e inyectarla en el cromatógrafo de gases. Alternativamente, podemos muestrear el espacio de cabeza con un SPME.
La desorción térmica es un método útil para liberar analitos volátiles de sólidos. Colocamos una porción del sólido en un tubo de acero inoxidable revestido de vidrio. Después de purgar con gas portador para eliminar cualquier O 2 que pudiera estar presente, calentamos la muestra. Los analitos volátiles son barridos del tubo por un gas inerte y transportados al GC. Debido a que la volatilización no es un proceso rápido, los analitos volátiles a menudo se concentran en la parte superior de la columna enfriando la entrada de la columna por debajo de la temperatura ambiente, un proceso conocido como enfoque criogénico. Una vez completada la volatilización, la entrada de la columna se calienta rápidamente, liberando los analitos para que viajen a través de la columna.
La razón para eliminar O 2 es evitar que la muestra sufra una reacción de oxidación cuando se calienta.
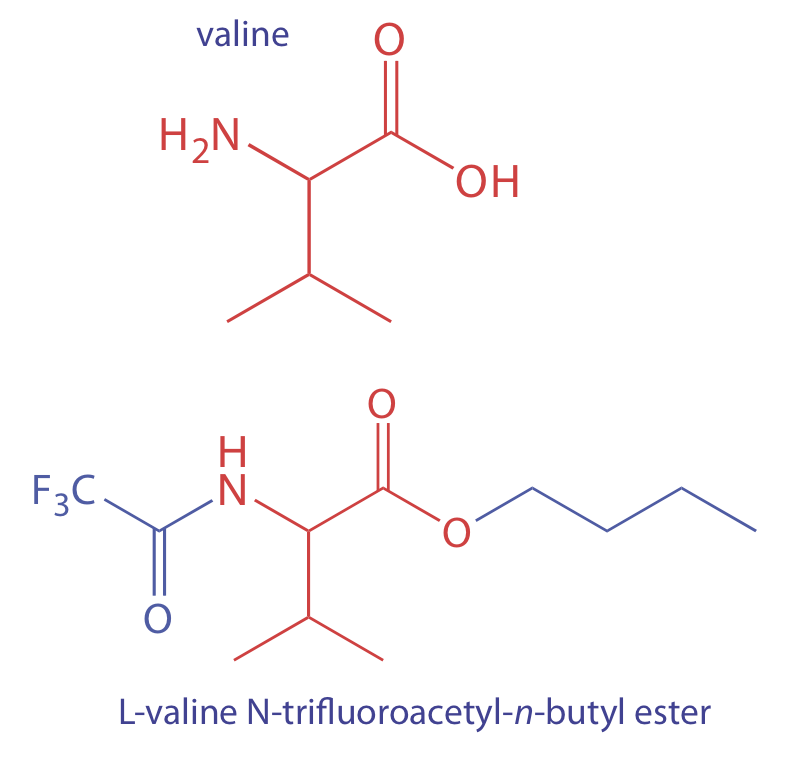
Para analizar un analito no volátil debemos convertirlo en una forma volátil. Por ejemplo, los aminoácidos no son lo suficientemente volátiles para analizarlos directamente por cromatografía de gases. La reacción de un aminoácido, como la valina, con 1-butanol y cloruro de acetilo produce un aminoácido esterificado. El tratamiento posterior con ácido trifluoroacético da el derivado volátil del éster N-trifluoroacetil- n-butílico del aminoácido.
Ajuste de la concentración del analito
En un analito la concentración es demasiado pequeña para dar una señal adecuada, entonces debemos concentrar el analito antes de inyectar la muestra en el cromatógrafo de gases. Un beneficio secundario de muchos métodos de extracción es que a menudo concentran los analitos. Los materiales orgánicos volátiles aislados de una muestra acuosa mediante una purga y trampa, por ejemplo, se concentran tanto como\(1000 \times\).
Si un analito está demasiado concentrado, es fácil sobrecargar la columna, dando como resultado un frente de pico (ver Figura 12.2.7) y una mala separación. Además, la concentración del analito puede exceder la respuesta lineal del detector. Inyectar menos muestra o diluir la muestra con un disolvente volátil, como el cloruro de metileno, son dos posibles soluciones a este problema.
Inyección de la muestra
En el Capítulo 12.3 examinamos varias explicaciones de por qué la banda de un soluto aumenta de ancho a medida que pasa por la columna, un proceso que llamamos ensanchamiento de banda. También introducimos una fuente adicional de ensanchamiento de banda si no conseguimos inyectar la muestra en el volumen mínimo posible de fase móvil. Hay dos fuentes principales de este ensanchamiento de banda precolumna: inyectar la muestra en una corriente móvil de fase móvil e inyectar una muestra líquida en lugar de una muestra gaseosa. El diseño del inyector de un cromatógrafo de gases ayuda a minimizar estos problemas.
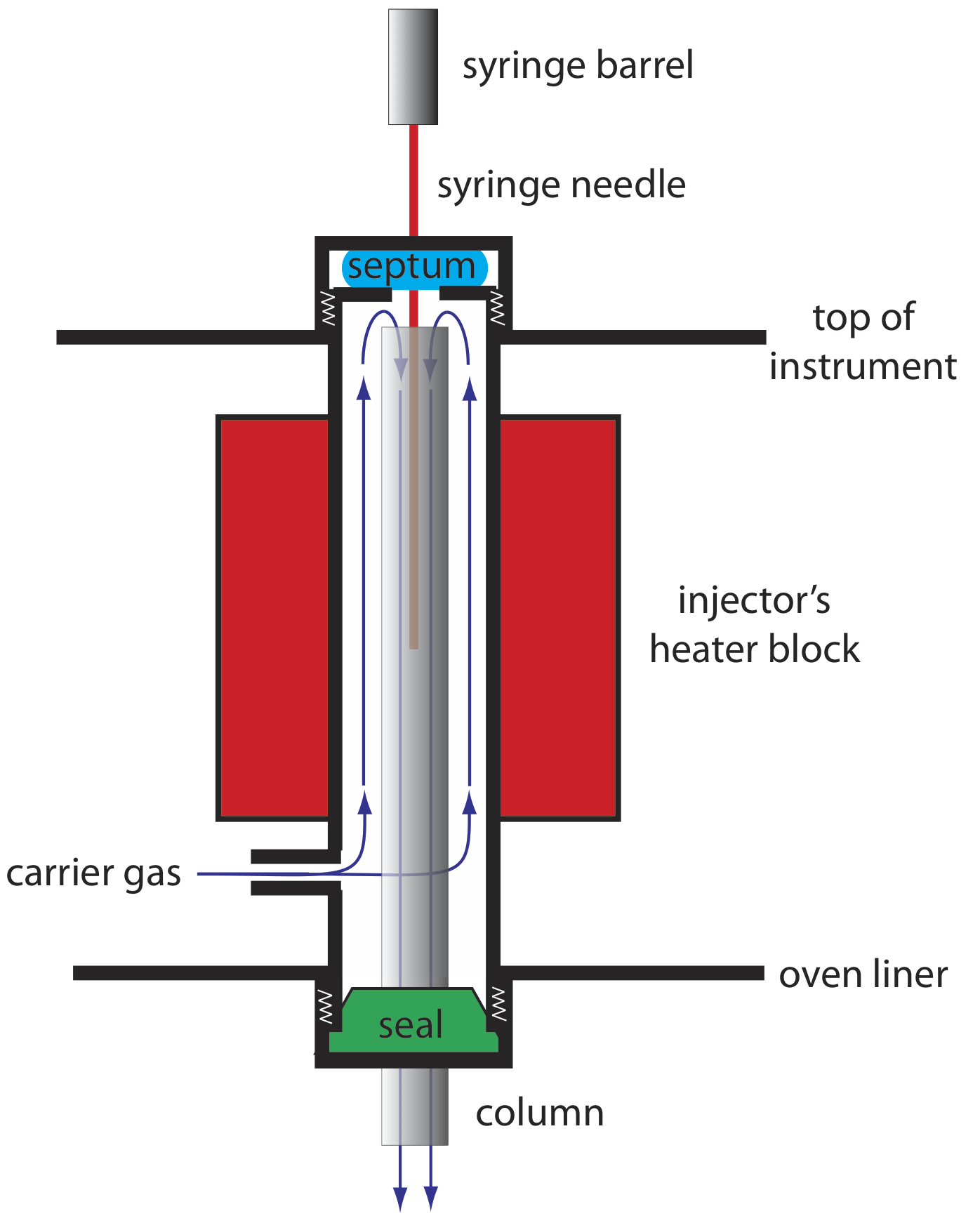
Un ejemplo de un puerto de inyección simple para una columna empaquetada se muestra en la Figura 12.4.7 . La parte superior de la columna encaja dentro de un bloque inyector calentado, con gas portador entrando desde la parte inferior. La muestra se inyecta a través de un tabique de goma usando una jeringa de microlitros, como la que se muestra en la Figura 12.4.8 . Inyectar la muestra directamente en la columna minimiza el ensanchamiento de banda porque mezcla la muestra con la menor cantidad posible de gas portador. El bloque inyector se calienta a una temperatura de al menos 50 o C por encima del punto de ebullición del soluto menos volátil, lo que asegura una rápida vaporización de los componentes de la muestra.

Debido a que el volumen de una columna capilar es significativamente menor que el de una columna empaquetada, requiere un estilo diferente de inyector para evitar sobrecargar la columna con muestra. La figura 12.4.9 muestra un diagrama esquemático de un inyector típico de split/splitless para su uso con una columna capilar.
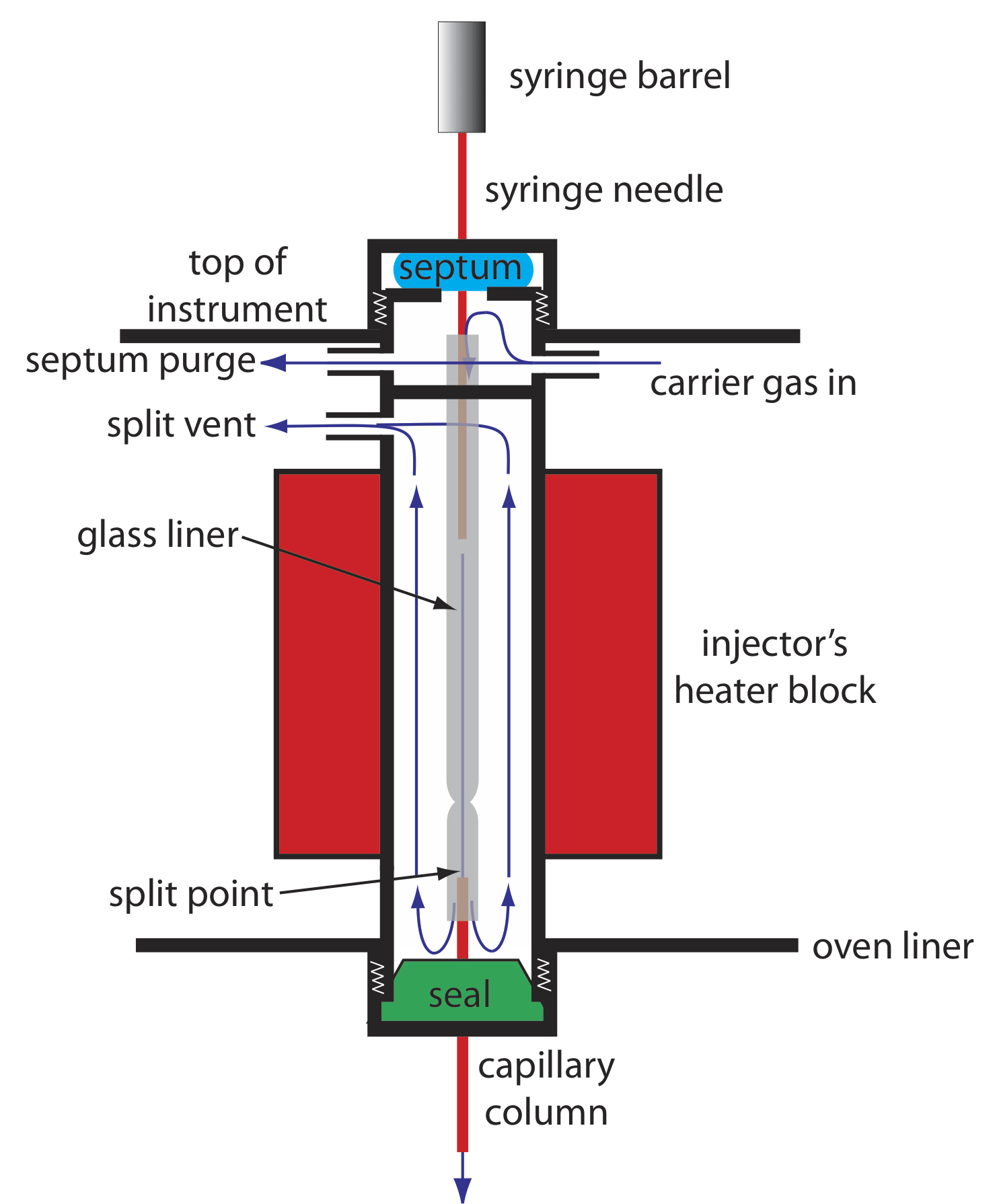
En una inyección dividida inyectamos la muestra a través de un septo de goma usando una jeringa de microlitro. En lugar de inyectar la muestra directamente en la columna, se inyecta en un revestimiento de vidrio donde se mezcla con el gas portador. En el punto de división, una pequeña fracción del gas portador y la muestra ingresa a la columna capilar con el resto saliendo por el respiradero dividido. Al controlar el caudal del gas portador a medida que ingresa al inyector, y su caudal a través de la purga del tabique y la ventilación dividida, podemos controlar la fracción de muestra que ingresa a la columna capilar, típicamente 0.1— 10%.
Por ejemplo, si el caudal de gas portador es de 50 ml/min, y los caudales para la purga del tabique y el respiradero dividido son de 2 ml/min y 47 ml/min, respectivamente, entonces el caudal a través de la columna es de 1 ml/min (= 50 — 2 — 47). La proporción de muestra que ingresa a la columna es 1/50, o 2%.
En una inyección sin división, que es útil para el análisis de trazas, cerramos el respiradero dividido y permitimos que todo el gas portador que pasa a través del revestimiento de vidrio ingrese a la columna, esto permite que prácticamente toda la muestra ingrese a la columna. Debido a que el caudal a través del inyector es bajo, el ensanchamiento significativo de la banda precolumna es un problema. El mantenimiento de la temperatura de la columna aproximadamente 20—25 o C por debajo del punto de ebullición del disolvente permite que el disolvente se condense a la entrada de la columna capilar, formando una barrera que atrapa los solutos. Después de permitir que los solutos se concentren, se incrementa la temperatura de la columna y se inicia la separación.
Para muestras que se descomponen fácilmente, puede ser necesaria una inyección en columna. En este método la muestra se inyecta directamente en la columna sin calentar. Luego se incrementa la temperatura de la columna, volatilizando la muestra con una temperatura tan baja como sea práctico.
Control de temperatura
El control de la temperatura de la columna es crítico para lograr una buena separación cuando se usa cromatografía de gases. Por esta razón la columna se coloca dentro de un horno termostático (ver Figura 12.4.1 ). En una separación isotérmica mantenemos la columna a una temperatura constante. Para aumentar la interacción entre los solutos y la fase estacionaria, la temperatura generalmente se establece ligeramente por debajo de la del soluto de menor punto de ebullición.
Una dificultad con una separación isotérmica es que una temperatura que favorece la separación de un soluto de bajo punto de ebullición puede conducir a un tiempo de retención inaceptablemente largo para un soluto de mayor punto de ebullición. La programación de temperatura proporciona una solución a este problema. Al inicio del análisis establecemos la temperatura inicial de la columna por debajo de la del soluto de menor punto de ebullición. A medida que avanza la separación, aumentamos lentamente la temperatura ya sea a una velocidad uniforme o en una serie de pasos.
Detectores para cromatografía de gases
La parte final de un cromatógrafo de gases es el detector. El detector ideal tiene varias características deseables: un bajo límite de detección, una respuesta lineal en un amplio rango de concentraciones de solutos (lo que facilita el trabajo cuantitativo), sensibilidad para todos los solutos o selectividad para una clase específica de solutos, y una insensibilidad a un cambio en el caudal o temperatura.
Detector de Conductividad Térmica (TCD)
Uno de los primeros detectores de cromatografía de gases aprovecha la conductividad térmica de la fase móvil. A medida que la fase móvil sale de la columna pasa sobre un filamento de alambre de tungsteno-renio (ver Figura 12.4.10 . La resistencia eléctrica del filamento depende de su temperatura, la cual, a su vez, depende de la conductividad térmica de la fase móvil. Debido a su alta conductividad térmica, el helio es la fase móvil de elección cuando se utiliza un detector de conductividad térmica (TCD).
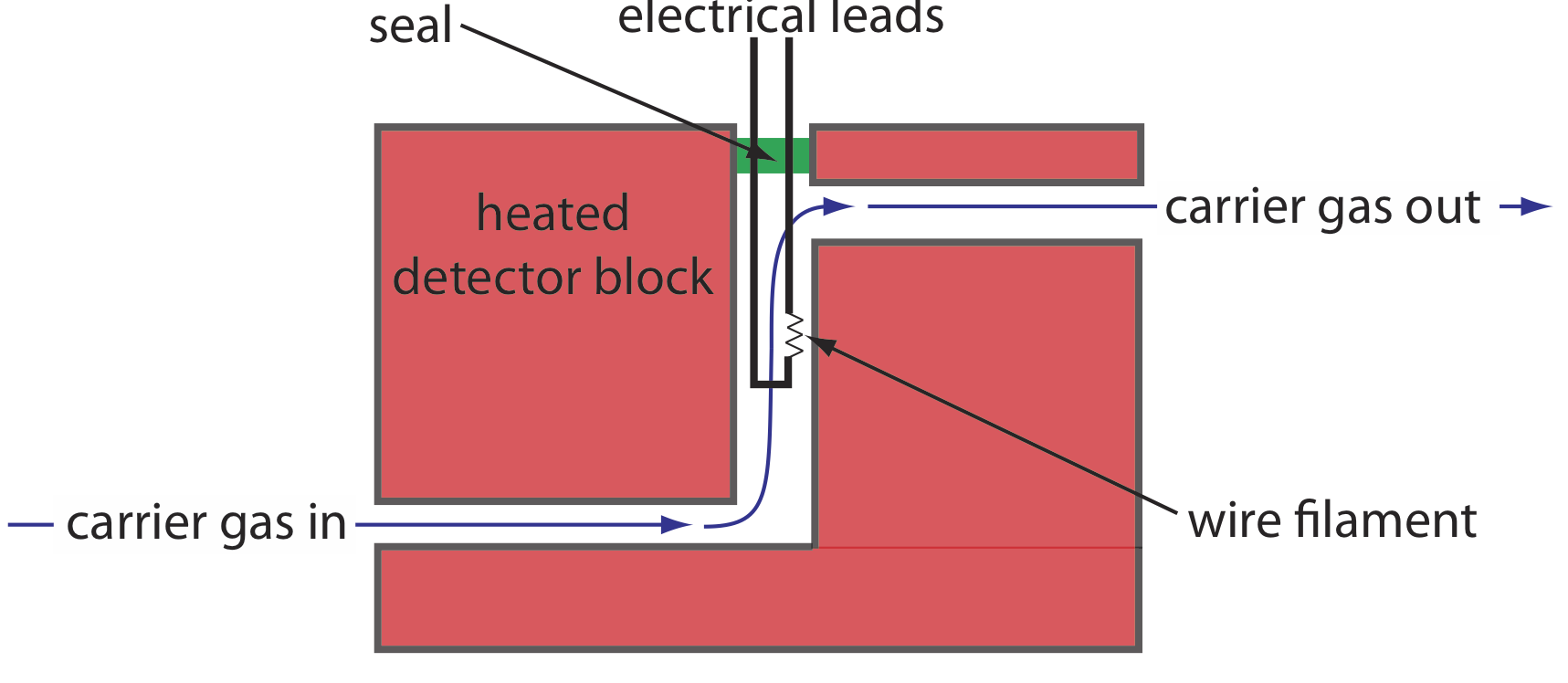
La conductividad térmica, como su nombre indica, es una medida de la facilidad con la que una sustancia conduce el calor. Un gas con una alta conductividad térmica aleja el calor del filamento y, por lo tanto, lo enfría más rápidamente que un gas con baja conductividad térmica.
Cuando un soluto eluye de la columna, la conductividad térmica de la fase móvil en la celda TCD disminuye y la temperatura del filamento de alambre, y por lo tanto su resistencia, aumenta. Una celda de referencia, a través de la cual solo pasa la fase móvil, corrige cualquier variación dependiente del tiempo en el caudal, la presión o la energía eléctrica, todas las cuales afectan la resistencia del filamento.
Debido a que todos los solutos afectan la conductividad térmica de la fase móvil, el detector de conductividad térmica es un detector universal. Otra ventaja es la respuesta lineal del TCD sobre un rango de concentración que abarca 10 4 —10 5 órdenes de magnitud. El detector también es no destructivo, lo que nos permite recuperar analitos usando una trampa fría posdetector. Una desventaja significativa del detector de TCD es su escaso límite de detección para la mayoría de los analitos.
Detector de Ionización de Llama (FID)
La combustión de un compuesto orgánico en una llama H 2/aire da como resultado una llama que contiene electrones y cationes orgánicos, presumiblemente CHO +. La aplicación de un potencial de aproximadamente 300 voltios a través de la llama crea una pequeña corriente de aproximadamente 10 —9 a 10 —12 amperios. Cuando se amplifica, esta corriente proporciona una señal analítica útil. Esta es la base del popular detector de ionización de llama, un diagrama esquemático del cual se muestra en la Figura 12.4.11 .
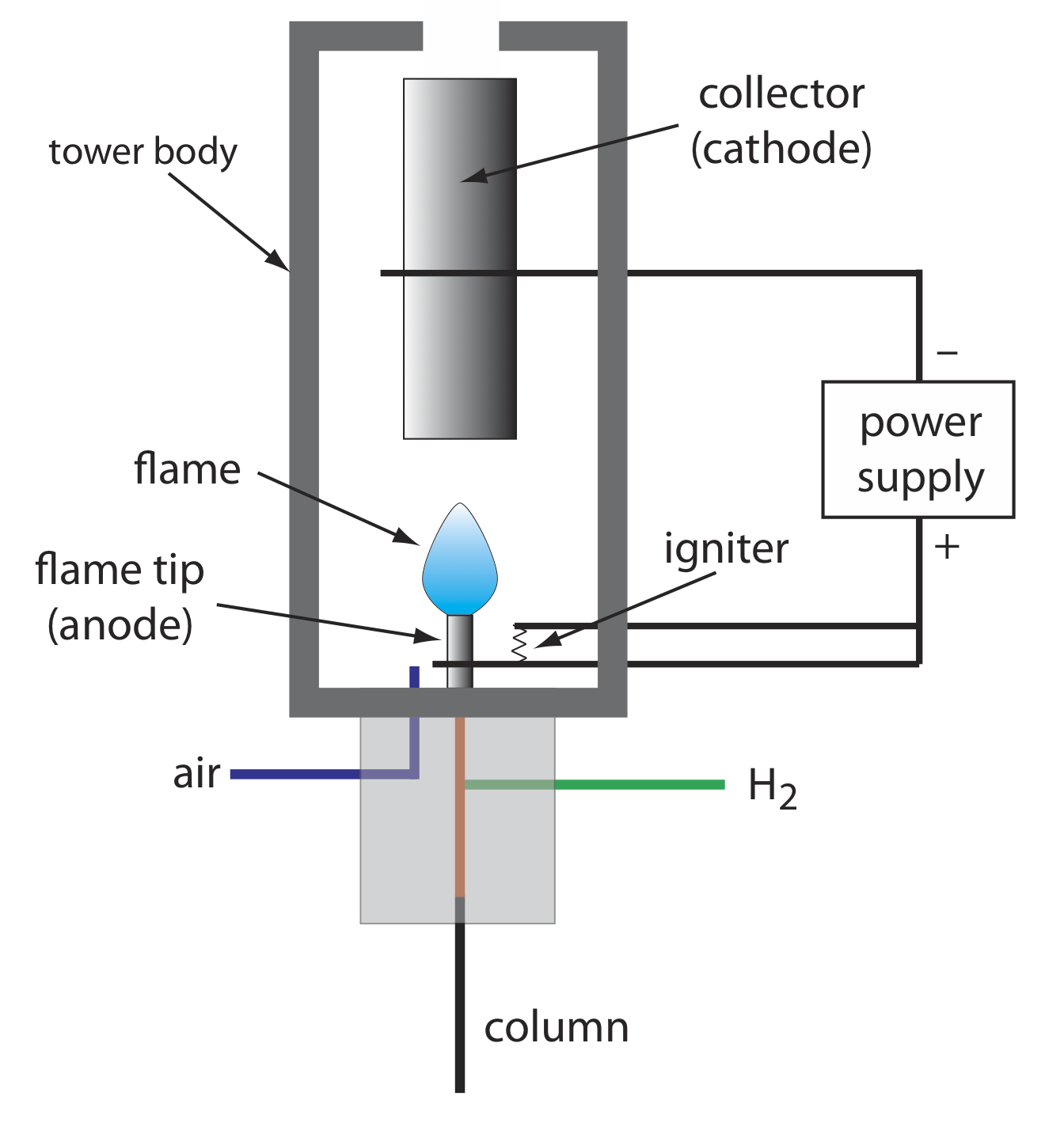
La mayoría de los átomos de carbono, excepto los de los grupos carbonilo y carboxílico, generan una señal, lo que convierte al FID en un detector casi universal para compuestos orgánicos. La mayoría de los compuestos inorgánicos y muchos gases, como H 2 O y CO 2, no se detectan, lo que convierte al detector FID en un detector útil para el análisis de analitos orgánicos en muestras ambientales atmosféricas y acuosas. Las ventajas del FID incluyen un límite de detección que es aproximadamente de dos a tres órdenes de magnitud menor que el de un detector de conductividad térmica, y una respuesta lineal superior a 10 6 —10 7 órdenes de magnitud en la cantidad de analito inyectado. La muestra, por supuesto, se destruye cuando se utiliza un detector de ionización de llama.
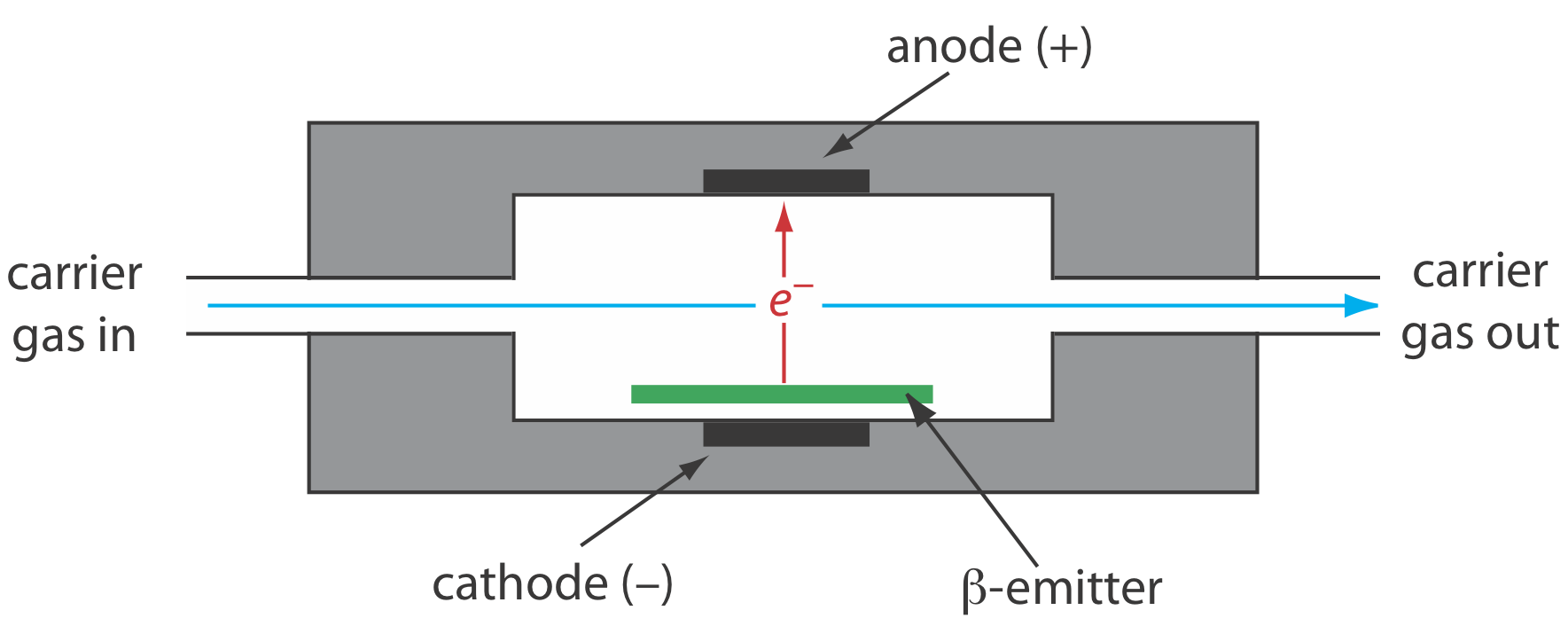
Detector de captura de electrones (ECD)
El detector de captura de electrones es un ejemplo de un detector selectivo. Como se muestra en la Figura 12.4.12 , el detector consiste en un\(\beta\) -emisor, tal como 63 Ni. Los electrones emitidos ionizan la fase móvil, generalmente N 2, generando una corriente estacionaria entre un par de electrodos. Cuando un soluto con una alta afinidad para capturar electrones eluye de la columna, la corriente disminuye, lo que sirve como señal. El ECD es altamente selectivo hacia solutos con grupos funcionales electronegativos, como halógenos y grupos nitro, y es relativamente insensible a aminas, alcoholes e hidrocarburos. Aunque su límite de detección es excelente, su rango lineal se extiende solo en aproximadamente dos órdenes de magnitud.
Una\(\beta\) partícula es un electrón.
Espectrómetro de masas (MS)
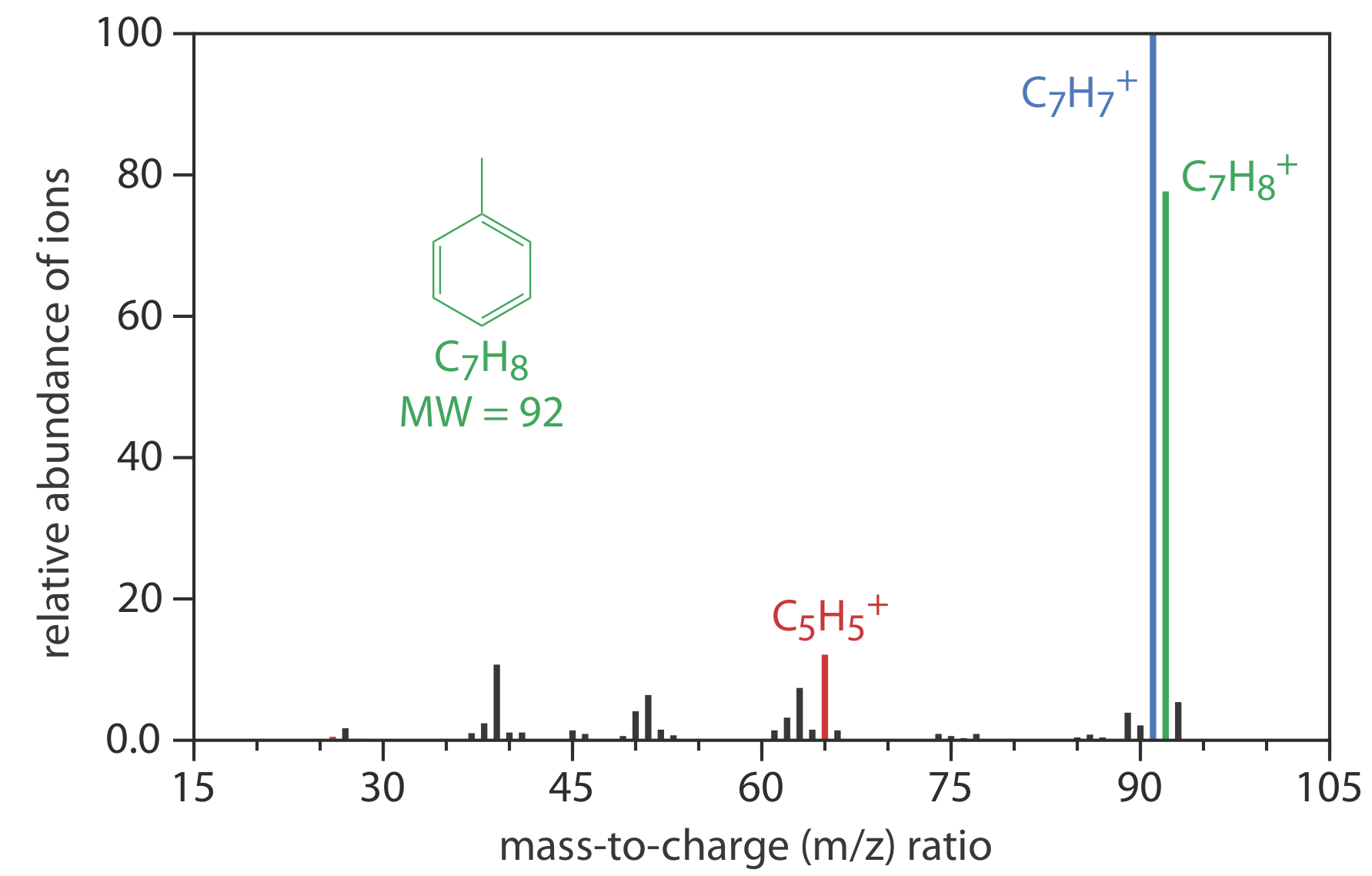
Un espectrómetro de masas es un instrumento que ioniza una molécula gaseosa utilizando suficiente energía para que el ion resultante se descomponga en iones más pequeños. Debido a que estos iones tienen diferentes relaciones de masa a carga, es posible separarlos usando un campo magnético o un campo eléctrico. El espectro de masas resultante contiene información tanto cuantitativa como cualitativa sobre el analito. La Figura 12.4.13 muestra un espectro de masas para tolueno.
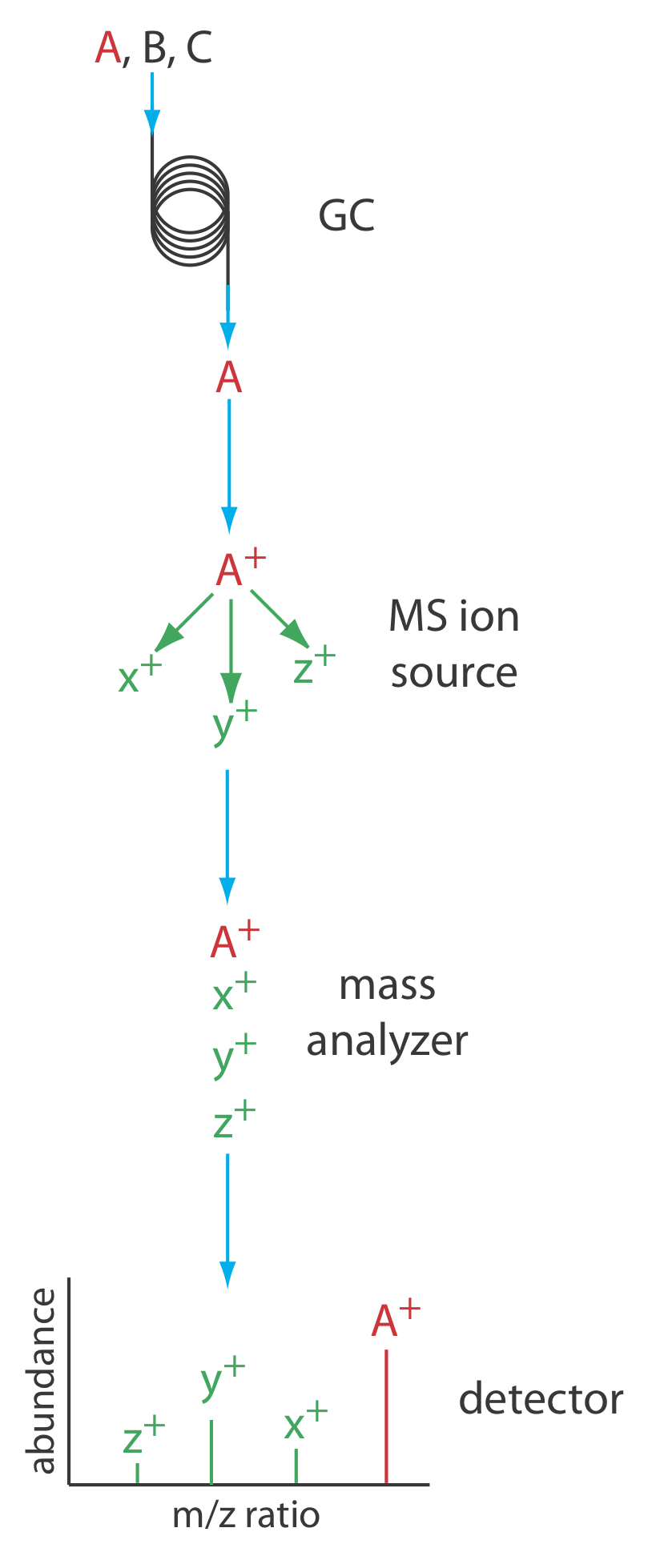
La Figura 12.4.14 muestra un diagrama de bloques de un instrumento típico de cromatografía de gases-espectrómetro de masas (GC-MS). El efluente de la columna ingresa a la fuente de iones del espectrómetro de masas de manera que elimina la mayoría del gas portador. En la cámara de ionización, las moléculas restantes, una mezcla de gas portador, disolvente y solutos, experimentan ionización y fragmentación. El analizador de masas del espectrómetro de masas separa los iones por su relación masa-carga y un detector cuenta los iones y muestra el espectro de masas.
Existen varias opciones para monitorear un cromatograma cuando se usa un espectrómetro de masas como detector. El método más común es escanear continuamente todo el espectro de masas y reportar la señal total para todos los iones que llegan al detector durante cada exploración. Este escaneo de iones totales proporciona detección universal para todos los analitos. Podemos lograr cierto grado de selectividad monitoreando una o más relaciones específicas de masa a carga, un proceso llamado monitoreo de iones selectivos. Un espectrómetro de masas proporciona excelentes límites de detección, típicamente de 25 fg a 100 pg, con un rango lineal de 10 5 órdenes de magnitud. Debido a que registramos continuamente el espectro de masas del eluyente de la columna, podemos retroceder y examinar el espectro de masas para cualquier incremento de tiempo. Esta es una clara ventaja para GC-MS porque podemos usar el espectro de masas para ayudar a identificar los componentes de una mezcla.
Para más detalles sobre espectrometría de masas consulte Introducción a la espectrometría de masas de Michael Samide y Olujide Akinbo, un recurso que forma parte de la Biblioteca Digital de Ciencias Analíticas.
Otros Detectores
Dos detectores adicionales son similares en diseño a un detector de ionización de llama. En el detector fotométrico de llama, la emisión óptica de fósforo y azufre proporciona un detector selectivo para compuestos que contienen estos elementos. El detector termiónico responde a compuestos que contienen nitrógeno o fósforo.
Un espectrofotómetro infrarrojo por transformada de Fourier (FT—IR) también puede servir como detector. En GC-FT—IR, el efluente de la columna fluye a través de una celda óptica construida a partir de un tubo Pyrex de 10—40 cm con un diámetro interno de 1—3 mm. La superficie interior de la celda está recubierta con una capa reflectante de oro. Las múltiples reflexiones de la radiación fuente a medida que se transmite a través de la célula aumentan la longitud de la trayectoria óptica a través de la muestra. Como es el caso de GC-MS, un detector FT—IR registra continuamente el espectro del eluyente de la columna, lo que nos permite examinar el espectro IR para cualquier incremento de tiempo.
Consulte la Sección 10.3 para una discusión sobre espectroscopia e instrumentación FT-IR.
Aplicaciones Cuantitativas
La cromatografía de gases es ampliamente utilizada para el análisis de una amplia gama de muestras en laboratorios ambientales, clínicos, farmacéuticos, bioquímicos, forenses, de ciencias de los alimentos y petroquímicos. Table 12.4.2 proporciona algunos ejemplos representativos de aplicaciones.
Cálculos cuantitativos
En un análisis GC el área bajo el pico es proporcional a la cantidad de analito inyectado en la columna. El área de un pico está determinada por la integración, que generalmente es manejada por la computadora del instrumento o por una grabadora electrónica integradora. Si dos picos se resuelven completamente, la determinación de sus respectivas áreas es sencilla.
Antes de integrar grabadoras electrónicas y computadoras, se utilizaron dos métodos para encontrar el área bajo una curva. Un método utilizó un planímetro manual; al usar el planímetro para trazar el perímetro de un objeto, registra el área. Un segundo enfoque para encontrar el área de un pico es el método de cortar y pesar. El cromatograma se registra en una hoja de papel y cada pico de interés se corta y se pesa. Suponiendo que el papel es uniforme en grosor y densidad de fibras, la relación de pesos para dos picos es la misma que la relación de áreas. Por supuesto, este enfoque destruye tu cromatograma.
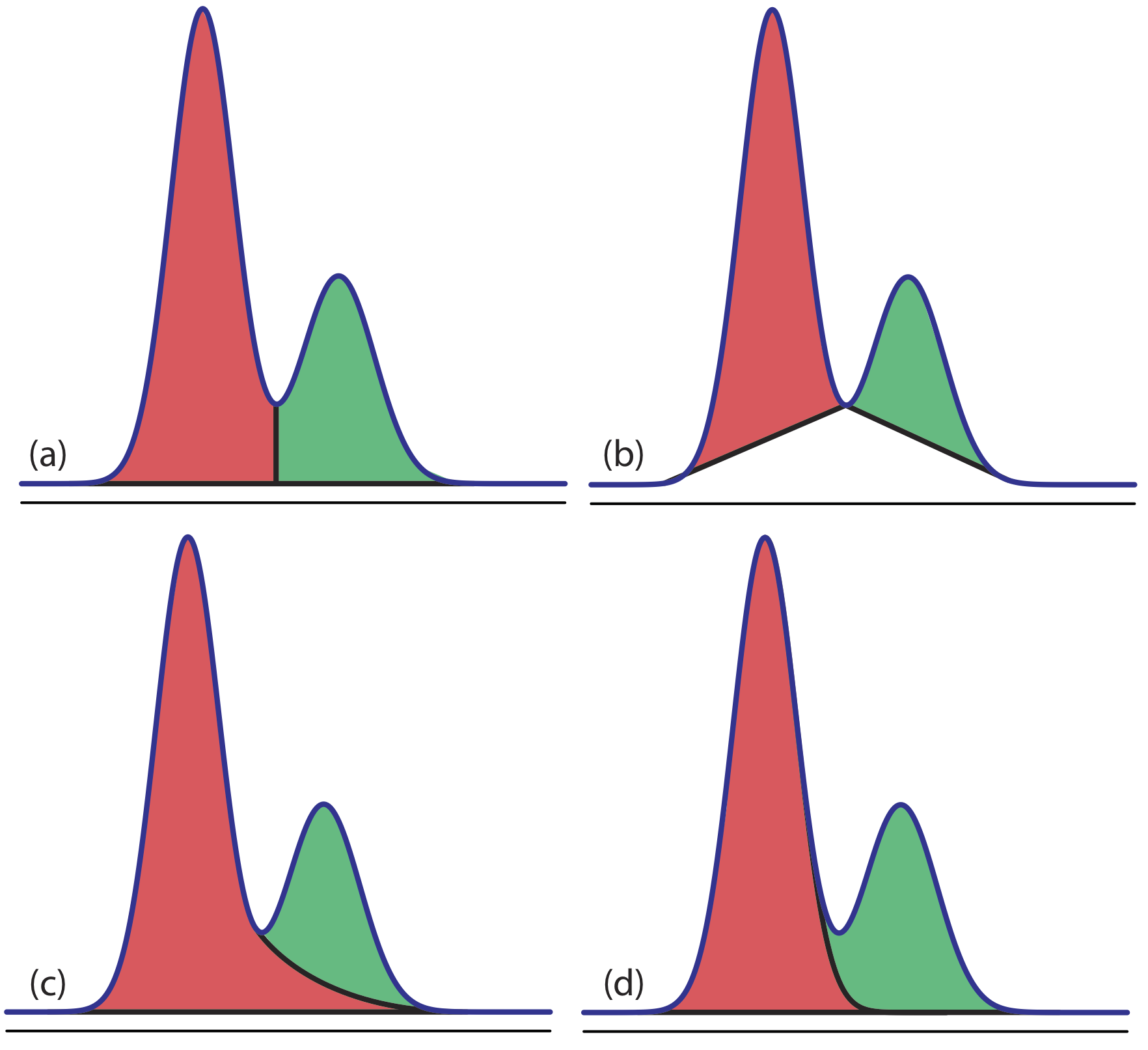
Los picos superpuestos, sin embargo, requieren una elección entre una de varias opciones para dividir el área compartida por los dos picos (Figura 12.4.15 ). El método que utilizamos depende del tamaño relativo de los dos picos y su resolución. En algunos casos, el uso de alturas de pico proporciona resultados más precisos [(a) Bicking, M. K. L. Chromatography Online, abril de 2006; (b) Bicking, M. K. L. Chromatography Online, junio de 2006].
Para el trabajo cuantitativo necesitamos establecer una curva de calibración que relacione la respuesta del detector con la concentración del analito. Si el volumen de inyección es idéntico para cada estándar y muestra, entonces una estandarización externa proporciona resultados precisos y precisos. Desafortunadamente, incluso en las mejores condiciones la precisión relativa de las inyecciones replicadas puede diferir en un 5%; a menudo es sustancialmente peor. Para trabajos cuantitativos que requieran alta precisión y precisión, se recomienda el uso de estándares internos.
Para revisar el método de las normas internas, véase el Capítulo 5.3.
Marriott y Carpenter reportan los siguientes datos para cinco inyecciones replicadas de una mezcla que contiene 1% v/v de metil isobutil cetona y 1% v/v de p-xileno en diclorometano [Marriott, P. J.; Carpenter, P. D. J. Chem. Educ. 1996, 73, 96—99].
| inyección | pico | área de pico (unidades arb.) |
|---|---|---|
| I | 1 | 48075 |
| 2 | 78112 | |
| II | 1 | 85829 |
| 2 | 135404 | |
| III | 1 | 84136 |
| 2 | 132332 | |
| IV | 1 | 71681 |
| 2 | 112889 | |
| V | 1 | 58054 |
| 2 | 91287 |
Supongamos que p-xileno (pico 2) es el analito, y que la metil isobutil cetona (pico 1) es el estándar interno. Determinar el intervalo de confianza del 95% para una estandarización de punto único con y sin usar el estándar interno.
Solución
Para una estandarización externa de un solo punto ignoramos el estándar interno y determinamos la relación entre el área del pico para p-xileno, A 2, y la concentración, C 2, de p-xileno.
\[A_{2}=k C_{2} \nonumber\]
Sustituyendo la concentración conocida por p-xileno (1% v/v) y las áreas de pico apropiadas, da los siguientes valores para la constante k.
\[78112 \quad 135404 \quad 132332 \quad 112889 \quad 91287 \nonumber\]
El valor promedio para k es de 110 000 con una desviación estándar de 25 100 (una desviación estándar relativa de 22.8%). El intervalo de confianza del 95% es
\[\mu=\overline{X} \pm \frac{t s}{\sqrt{n}}=111000 \pm \frac{(2.78)(25100)}{\sqrt{5}}=111000 \pm 31200 \nonumber\]
Para una estandarización interna, la relación entre el área del pico del analito, A2, el área del pico del patrón interno, A1, y sus respectivas concentraciones, C 2 y C1, es
\[\frac{A_{2}}{A_{1}}=k \frac{C_{2}}{C_{1}} \nonumber\]
Sustituyendo en las concentraciones conocidas y las áreas de pico apropiadas da los siguientes valores para la constante k.
\[1.5917 \quad 1.5776 \quad 1.5728 \quad 1.5749 \quad 1.5724 \nonumber\]
El valor promedio para k es de 1.5779 con una desviación estándar de 0.0080 (una desviación estándar relativa de 0.507%). El intervalo de confianza del 95% es
\[\mu=\overline{X} \pm \frac{t s}{\sqrt{n}}=1.5779 \pm \frac{(2.78)(0.0080)}{\sqrt{5}}=1.5779 \pm 0.0099 \nonumber\]
Aunque existe una variación sustancial en las áreas de pico individuales para este conjunto de inyecciones replicadas, el estándar interno compensa estas variaciones, proporcionando una calibración más precisa y precisa.
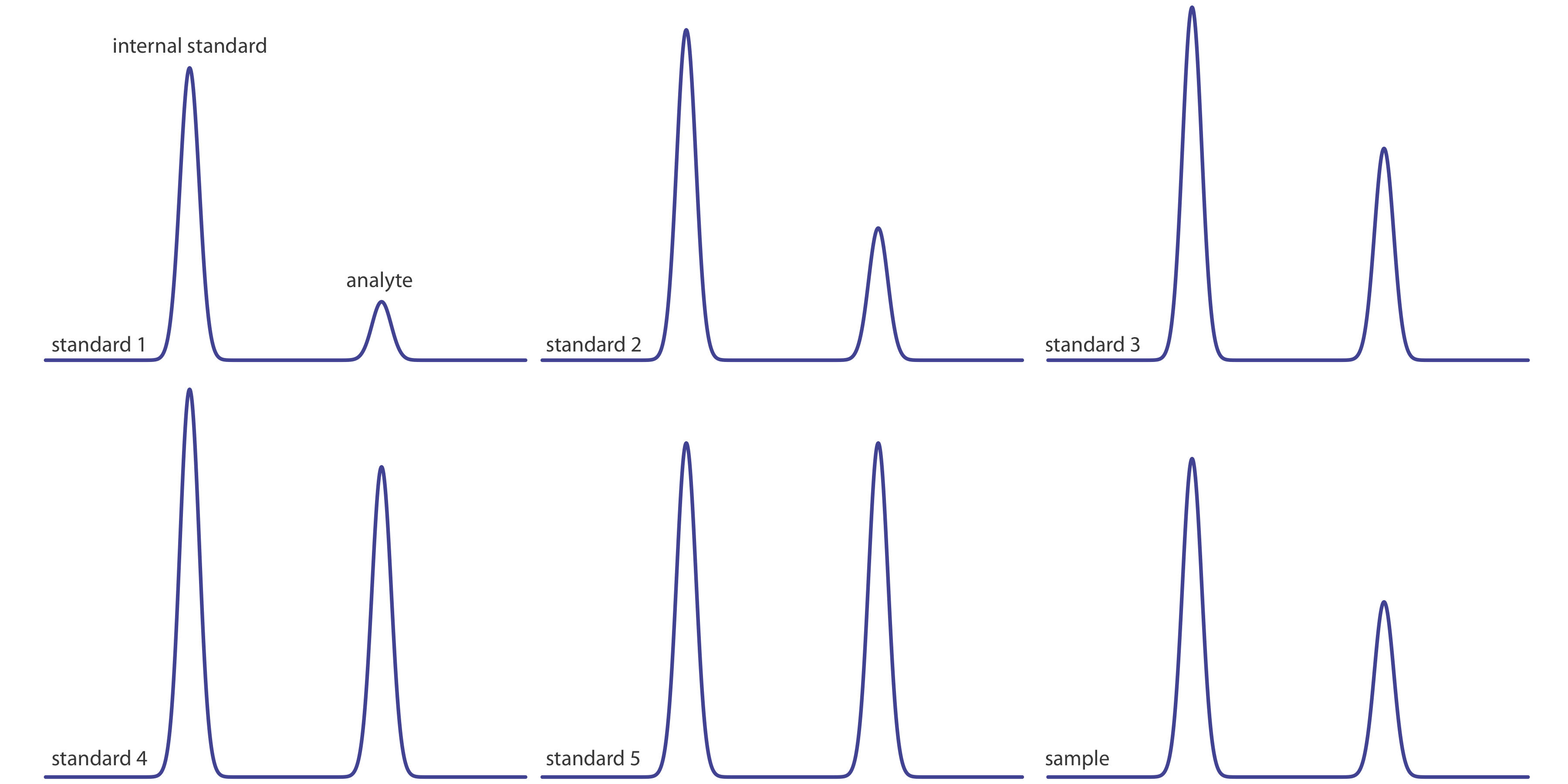
La Figura 12.4.16 muestra cromatogramas para cinco patrones y para una muestra. Cada patrón y muestra contiene la misma concentración de un patrón interno, que es de 2.50 mg/mL. Para los cinco estándares, las concentraciones de analito son 0.20 mg/mL, 0.40 mg/mL, 0.60 mg/mL, 0.80 mg/mL y 1.00 mg/mL, respectivamente. Determinar la concentración de analito en la muestra (a) ignorando los estándares internos y creando una curva de calibración de estándares externos, y (b) creando una curva de calibración estándar interna. Para cada abordaje, reportar la concentración del analito y el intervalo de confianza del 95%. Use alturas de pico en lugar de áreas de picos.
- Responder
-
En la siguiente tabla se resumen mis medidas de las alturas de los picos para cada estándar y la muestra, y su relación (aunque sus valores absolutos para alturas de pico diferirán de los míos, dependiendo del tamaño de su monitor o impresión, sus relaciones relativas de altura máxima deben ser similares a las mías).
[estándar] (mg/mL) altura de pico estándar (mm) altura pico del analito (mm) relación de altura pico 0.20
35
7 0.20 0.40 41 16 0.39 0.60 44 27 0.61 0.80 48 39 0.81 1.00 41 41 1.00 muestra 39 21 0.54 La figura (a) muestra la curva de calibración y la ecuación de calibración cuando ignoramos el estándar interno. Sustituyendo la altura del pico de la muestra en la ecuación de calibración da la concentración del analito en la muestra como 0.49 mg/mL. El intervalo de confianza del 95% es de ±0.24 mg/mL. La curva de calibración muestra bastante dispersión en los datos debido a la incertidumbre en los volúmenes de inyección.
La Figura (b) muestra la curva de calibración y la ecuación de calibración cuando se incluye el estándar interno. Sustituir la relación de altura máxima de la muestra en la ecuación de calibración da la concentración del analito en la muestra como 0.54 mg/mL. El intervalo de confianza del 95% es de ±0.04 mg/mL.
Para revisar el uso de Excel o R para cálculos de regresión e intervalos de confianza, consulte el Capítulo 5.5.

Los datos para este ejercicio se crearon de manera que la concentración real del analito sea de 0.55 mg/mL. Dada la resolución de la escala de mi gobernante, mi respuesta es bastante razonable. Sus medidas pueden ser ligeramente diferentes, pero sus respuestas deben estar cerca de los valores reales.
Aplicaciones Cualitativas
Además de un análisis cuantitativo, también podemos utilizar la cromatografía para identificar los componentes de una mezcla. Como se señaló anteriormente, cuando se usa un FT—IR o un espectrómetro de masas como detector tenemos acceso al espectro completo del eluyente para cualquier tiempo de retención. Interpretando el espectro o buscando contra una biblioteca de espectros, podemos identificar el analito responsable de cada pico cromatográfico.
Además de identificar el componente responsable de un pico cromatográfico particular, también podemos usar los espectros guardados para evaluar la pureza máxima. Si solo un componente es responsable de un pico cromatográfico, entonces los espectros deben ser idénticos a lo largo de la elución del pico. Si un espectro al comienzo de la elución del pico es diferente de un espectro tomado cerca del final de la elución del pico, entonces al menos dos componentes están coeluyendo.
Cuando se utiliza un detector no espectroscópico, como un detector de ionización de llama, debemos encontrar otro enfoque si queremos identificar los componentes de una mezcla. Un enfoque es pinchar una muestra con el compuesto sospechoso y buscar un aumento en la altura del pico. También podemos comparar el tiempo de retención de un pico con el tiempo de retención para un compuesto conocido si usamos condiciones de operación idénticas.
Debido a que no es probable que los tiempos de retención de un compuesto en dos columnas idénticas sean los mismos, las diferencias en la eficiencia del empaque, por ejemplo, afectarán el tiempo de retención de un soluto en una columna empaquetada, no es posible crear una tabla de tiempos de retención estándar. El índice de retención de Kovat proporciona una solución al problema de igualar los tiempos de retención. En condiciones isotérmicas, los tiempos de retención ajustados para alcanos normales aumentan logarítmicamente. Kovat definió el índice de retención, I, para un alcano normal como 100 veces el número de átomos de carbono. Por ejemplo, el índice de retención es de 400 para butano, C 4 H 10 y 500 para pentano, C 5 H 12. Para determinar el índice de retención de un compuesto, I cpd, se utiliza la siguiente fórmula
donde\(t_{r,cpd}^{\prime}\) está el tiempo de retención ajustado del compuesto,\(t_{r,x}^{\prime}\) y\(t_{r,x+1}^{\prime}\) son los tiempos de retención ajustados para los alcanos normales que eluyen inmediatamente antes del compuesto e inmediatamente después del compuesto, respectivamente, e I x es el índice de retención para la normal alcano que eluye inmediatamente antes del compuesto. El índice de retención de un compuesto para un conjunto particular de condiciones cromatográficas (fase estacionaria, fase móvil, tipo de columna, longitud de columna, temperatura, etc.) es razonablemente consistente de día a día y entre diferentes columnas e instrumentos.
Se encuentran disponibles tablas de los índices de retención de Kovat; véase, por ejemplo, el Webbook de Química del NIST. La búsqueda de tolueno devuelve 341 valores de I para más de 20 fases estacionarias diferentes, y para columnas empaquetadas y columnas capilares.
En una separación de una mezcla de hidrocarburos se miden los siguientes tiempos de retención ajustados: 2.23 min para propano, 5.71 min para isobutano y 6.67 min para butano. ¿Cuál es el índice de retención de Kovat para cada uno de estos hidrocarburos?
Solución
El índice de retención de Kovat para un alcano normal es 100 veces el número de carbonos; así, para propano, I = 300 y para butano, I = 400. Para encontrar el índice de retención de Kovat para isobutano utilizamos la Ecuación\ ref {12.1}.
\[I_\text{isobutane} =100 \times \frac{\log (5.71)-\log (2.23)}{\log (6.67)-\log (2.23)}+300=386 \nonumber\]
Al usar una columna con la misma fase estacionaria que en el Ejemplo 12.4.2 , se encuentra que los tiempos de retención para propano y butano son de 4.78 min y 6.86 min, respectivamente. ¿Cuál es el tiempo de retención esperado para el isobutano?
- Responder
-
Debido a que estamos usando la misma columna podemos suponer que el índice de retención de isobutano de 386 permanece sin cambios. Usando la ecuación\ ref {12.1}, tenemos
\[386=100 \times \frac{\log x-\log (4.78)}{\log (6.86)-\log (4.78)}+300 \nonumber\]
donde x es el tiempo de retención para isobutano. Resolviendo para x, encontramos que
\[0.86=\frac{\log x-\log (4.78)}{\log (6.86)-\log (4.78)} \nonumber\]
\[0.135=\log x-0.679 \nonumber\]
\[0.814=\log x \nonumber\]
\[x=6.52 \nonumber\]
el tiempo de retención para isobutano es de 6.5 min.
La mejor manera de apreciar los detalles teóricos y prácticos discutidos en esta sección es examinar cuidadosamente un método analítico típico. Si bien cada método es único, la siguiente descripción de la determinación de trihalometanos en el agua potable proporciona un ejemplo instructivo de un procedimiento típico. La descripción aquí se basa en un Método 6232B en Métodos Estándar para el Examen de Agua y Aguas Residuales, 20th Ed., American Public Health Association: Washing- ton, DC, 1998.
Método Representativo 12.4.1: Determinación de Trihalometanos en Agua Potable
Descripción del método
En la mayoría de las aguas cloradas se encuentran trihalometanos, como cloroformo, CHCl 3 y bromoformo, CHBr 3. Debido a que el cloroformo es un presunto carcinógeno, la determinación de trihalometanos en los suministros públicos de agua potable es de considerable importancia. En este método se aíslan los trihalometanos CHCl 3, ChBrCl 2, ChBr 2 Cl y ChBr 3 mediante extracción líquido-líquido con pentano y se determinan mediante un cromatógrafo de gases equipado con un detector de captura de electrones.
Procedimiento
Recolectar la muestra en un vial de vidrio de 40 ml equipado con una tapa de rosca forrada con un tabique con cara de TFE. Llena el vial hasta que se desborde, asegurando que no haya burbujas de aire. Agregar 25 mg de ácido ascórbico como agente reductor para apagar la producción posterior de trihalometanos. Selle el vial y almacene la muestra a 4 o C por no más de 14 días.
Preparar una solución madre estándar para cada trihalometano colocando 9.8 mL de metanol en un matraz aforado de 10 mL. Dejar reposar el matraz durante 10 min, o hasta que todas las superficies humedecidas con metanol estén secas. Pesar el matraz al nivel más cercano a ±0.1 mg. Usando una jeringa de 100-μL, agregue 2 o más gotas de trihalometano al matraz aforado, permitiendo que cada gota caiga directamente en el metanol. Volver a pesar el matraz antes de diluir a volumen y mezclar. Transfiera la solución a un vial de vidrio de 40 mL equipado con una tapa de rosca forrada de TFE y reporte la concentración en μg/mL. Almacene las soluciones de stock a —10 a —20 o C y lejos de la luz.
Preparar un estándar de trabajo multicomponente a partir de los estándares de stock haciendo diluciones apropiadas de la solución madre con metanol en un matraz aforado. Elija concentraciones para que los estándares de calibración (vea más abajo) no requieran más de 20 μL de estándar de trabajo por cada 100 mL de agua.
Usando el estándar de trabajo multicomponente, prepare al menos tres, pero preferiblemente 5—7 estándares de calibración. Al menos un estándar debe estar cerca del límite de detección y los estándares deben correlacionarse con la concentración esperada de trihalometanos en las muestras. Usando un matraz volumétrico apropiado, prepare los estándares inyectando al menos 10 μL del estándar de trabajo debajo de la superficie del agua y diluya al volumen. Mezcle suavemente cada estándar tres veces solamente. Deseche la solución en el cuello del matraz aforado y luego transfiera la solución restante a un vial de vidrio de 40 ml con una tapa de rosca forrada de TFE. Si el estándar tiene un espacio de cabeza, debe analizarse dentro de 1 hora; los estándares sin espacio de cabeza pueden mantenerse hasta por 24 horas.
Preparar un patrón interno disolviendo 1,2-dibromopentano en hexano. Agregar una cantidad suficiente de esta solución al pentano para dar una concentración final de 30 μg 1,2-dibromopentano/ l.
Para preparar los estándares de calibración y las muestras para su análisis, abra el vial con tapa de rosca y retire 5 mL de la solución. Recapitular el vial y pesar al ±0.1 mg más cercano. Añadir 2.00 mL de pentano (con el estándar interno) a cada vial y agitar vigorosamente por 1 min. Deje que las dos fases se separen durante 2 min y luego use una pipeta de vidrio para transferir al menos 1 mL del pentano (la fase superior) a un vial de muestra de 1.8 mL con tapa roscada equipado con un septo TFE, y almacene a 4 o C hasta que esté listo para inyectarlos en el GC. Después de vaciar, enjuagar y secar el vial original de la muestra, pesarlo al ± 0.1 mg más cercano y calcular el peso de la muestra a ±0.1 g. Si la densidad es de 1.0 g/mL, entonces el peso de la muestra es equivalente a su volumen.
Inyectar una alícuota de 1—5 μL de los extractos de pentano en un GC equipado con una columna de vidrio de 2 mm de diámetro interno y 2 m de largo empaquetada con una fase estacionaria de escualano al 10% sobre un material de empaque de 80/100 malla Chromosorb WAW. Operar la columna a 67 o C y un caudal de 25 mL/min.
Se puede usar una variedad de otras columnas. Otra opción, por ejemplo, es una columna de sílice fundida de 30 m con un diámetro interno de 0.32 mm y un recubrimiento de 1 µm de la fase estacionaria DB-1. Se utiliza un caudal lineal de 20 cm/s con el siguiente programa de temperatura: mantener durante 5 min a 35 o C; aumentar a 70 o C a 10 o C/min; aumentar a 200 o C a 20 o C/min.
Preguntas
1. Una simple extracción líquido-líquido rara vez extrae el 100% del analito. ¿Cómo explica este método las extracciones incompletas?
Debido a que utilizamos el mismo procedimiento de extracción para las muestras y los estándares, esperamos razonablemente que la eficiencia de extracción sea la misma para todas las muestras y estándares; por lo tanto, la cantidad relativa de analito en dos muestras o estándares cualesquiera no se ve afectada por una extracción incompleta.
2. Es probable que las muestras de agua contengan trazas de otros compuestos orgánicos, muchos de los cuales extraerán en pentano junto con los trihalometanos. Una columna corta y empaquetada, como la utilizada en este método, generalmente no hace un trabajo particularmente bueno para resolver picos cromatográficos. ¿Por qué no necesitamos preocuparnos por estos otros compuestos?
Un detector de captura de electrones responde únicamente a compuestos, como los trihalometanos, que tienen grupos funcionales electronegativos. Debido a que un detector de captura de electrones no responderá a la mayoría de los compuestos potenciales interferentes, el cromatograma tendrá relativamente pocos picos distintos de los de los trihalometanos y el estándar interno.
3. Predecir el orden en que los cuatro analitos eluyen de la columna GC.
El tiempo de retención debe seguir los puntos de ebullición del compuesto, eluyendo desde el punto de ebullición más bajo hasta los puntos de ebullición más altos. El orden de elución esperado es CHCl 3 (61.2 o C), CHCl 2 Br (90 o C), CHClBr 2 (119 o C) y ChBr 3 (149.1 o C).
4. Aunque el cloroformo es un analito, también es un interferente porque está presente en niveles traza en el aire. Cualquier cloroformo presente en el aire del laboratorio, por ejemplo, puede ingresar a la muestra difundiendo a través del tabique de silicio del vial de muestra. ¿Cómo podemos determinar si las muestras están contaminadas de esta manera?
Un blanco de muestra de agua libre de trihalometano se mantiene con las muestras en todo momento. Si el blanco de la muestra no muestra evidencia de cloroformo, entonces podemos asumir con seguridad que las muestras también están libres de contaminación.
5. ¿Por qué es necesario recolectar muestras sin un espacio de cabeza (una capa de aire que recubre el líquido) en el vial de muestra?
Debido a que los trihalometanos son volátiles, la presencia de un espacio de cabeza permite la pérdida de analito de la muestra al espacio superior, lo que resulta en un error determinado negativo.
6. Al preparar la solución madre para cada trihalometano, el procedimiento especifica que añadimos dos o más gotas del compuesto puro dejándolas caer en un matraz aforado que contiene metanol. Sin embargo, al preparar los estándares de calibración, el estándar de trabajo debe inyectarse debajo de la superficie del metanol. Explique el motivo de esta diferencia.
Al preparar una solución madre, la pérdida potencial del trihalometano volátil no es importante porque determinamos su concentración en peso después de agregarlo al metanol y diluirlo a volumen. Sin embargo, cuando preparamos el estándar de calibración, debemos asegurarnos de que la adición de trihalometano sea cuantitativa; así, lo inyectamos debajo de la superficie para evitar la pérdida potencial de analito.
Evaluación
Escala de Operación
La cromatografía de gases se utiliza para analizar analitos presentes en niveles que van desde componentes principales hasta ultratracia. Dependiendo del detector, las muestras con analitos mayores y menores pueden necesitar diluirse antes del análisis. Los detectores de conductividad térmica e ionización de llama pueden manejar mayores cantidades de analito; otros detectores, como un detector de captura de electrones o un espectrómetro de masas, requieren cantidades sustancialmente menores de analito. Aunque el volumen de inyección para cromatografía de gases es bastante pequeño, típicamente aproximadamente un microlitro, la cantidad de muestra disponible debe ser suficiente para que la inyección sea una submuestra representativa. Para un analito traza, la cantidad real de analito inyectado suele estar en el rango de picogramas. Usando el Método Representativo 12.4.1 como ejemplo, una inyección de 3.0-μL de 1 μg/L de CHCl 3 es equivalente a 15 pg de CHCl 3, asumiendo una eficiencia de extracción del 100%.
Precisión
La precisión de un método cromatográfico de gases varía sustancialmente de una muestra a otra. Para muestras rutinarias, las precisiones del 1— 5% son comunes. Para analitos presentes en niveles de concentración muy bajos, para muestras con matrices complejas o para muestras que requieren un procesamiento significativo antes del análisis, la precisión puede ser sustancialmente peor. En el análisis para trihalometanos descrito en el Método Representativo 12.4.1, por ejemplo, son posibles errores determinados de hasta ± 25%.
Precisión
La precisión de un análisis cromatográfico de gases incluye contribuciones de muestreo, preparación de muestras y el instrumento. La desviación estándar relativa debida al instrumento suele ser del 1— 5%, aunque puede ser significativamente mayor. Las principales limitaciones son el ruido del detector, que afecta la determinación del área del pico y la reproducibilidad de los volúmenes de inyección. En el trabajo cuantitativo, el uso de un estándar interno compensa cualquier variabilidad en los volúmenes de inyección.
Sensibilidad
En un análisis cromatográfico de gases, la sensibilidad está determinada por las características del detector. De particular importancia para el trabajo cuantitativo es el rango lineal del detector; es decir, el rango de concentraciones sobre el cual una curva de calibración es lineal. Los detectores con un amplio rango lineal, como el detector de conductividad térmica y el detector de ionización de llama, se pueden usar para analizar muestras en un amplio rango de concentraciones sin ajustar las condiciones de operación. Otros detectores, como el detector de captura de electrones, tienen un rango lineal mucho más estrecho.
Selectividad
Debido a que combina la separación con el análisis, los métodos cromatográficos proporcionan una excelente selectividad. Al ajustar las condiciones suele ser posible diseñar una separación para que los analitos eluyan por sí mismos, incluso cuando la mezcla es compleja. Se obtiene selectividad adicional mediante el uso de un detector, como el detector de captura de electrones, que no responde a todos los compuestos.
Tiempo, Costo y Equipo
El tiempo de análisis puede variar desde varios minutos para muestras que contienen solo unos pocos constituyentes, hasta más de una hora para muestras más complejas. La preparación preliminar de la muestra puede aumentar sustancialmente el tiempo de análisis. La instrumentación para cromatografía de gases varía en precio desde económico (unos pocos miles de dólares) hasta caro (>$50,000). Los modelos más caros están diseñados para columnas capilares, incluyen una variedad de opciones de inyección y utilizan detectores más sofisticados, como un espectrómetro de masas, o incluyen múltiples detectores. Las columnas empaquetadas suelen costar <$200, y el costo de una columna capilar suele ser de $300—$1000.