
12.5: Cromatografía líquida de alto rendimiento
- Page ID
- 75640

En cromatografía líquida de alta resolución (HPLC) inyectamos la muestra, que está en forma de solución, en una fase móvil líquida. La fase móvil transporta la muestra a través de una columna empaquetada o capilar que separa los componentes de la muestra en función de su capacidad de partición entre la fase móvil y la fase estacionaria. La Figura 12.5.1 muestra un ejemplo de un instrumento típico de HPLC, que tiene varios componentes clave: depósitos que almacenan la fase móvil; una bomba para empujar la fase móvil a través del sistema; un inyector para introducir la muestra; una columna para separar la muestra en sus partes componentes; y un detector para monitorear el eluyente a medida que sale de la columna. Consideremos cada uno de estos componentes.
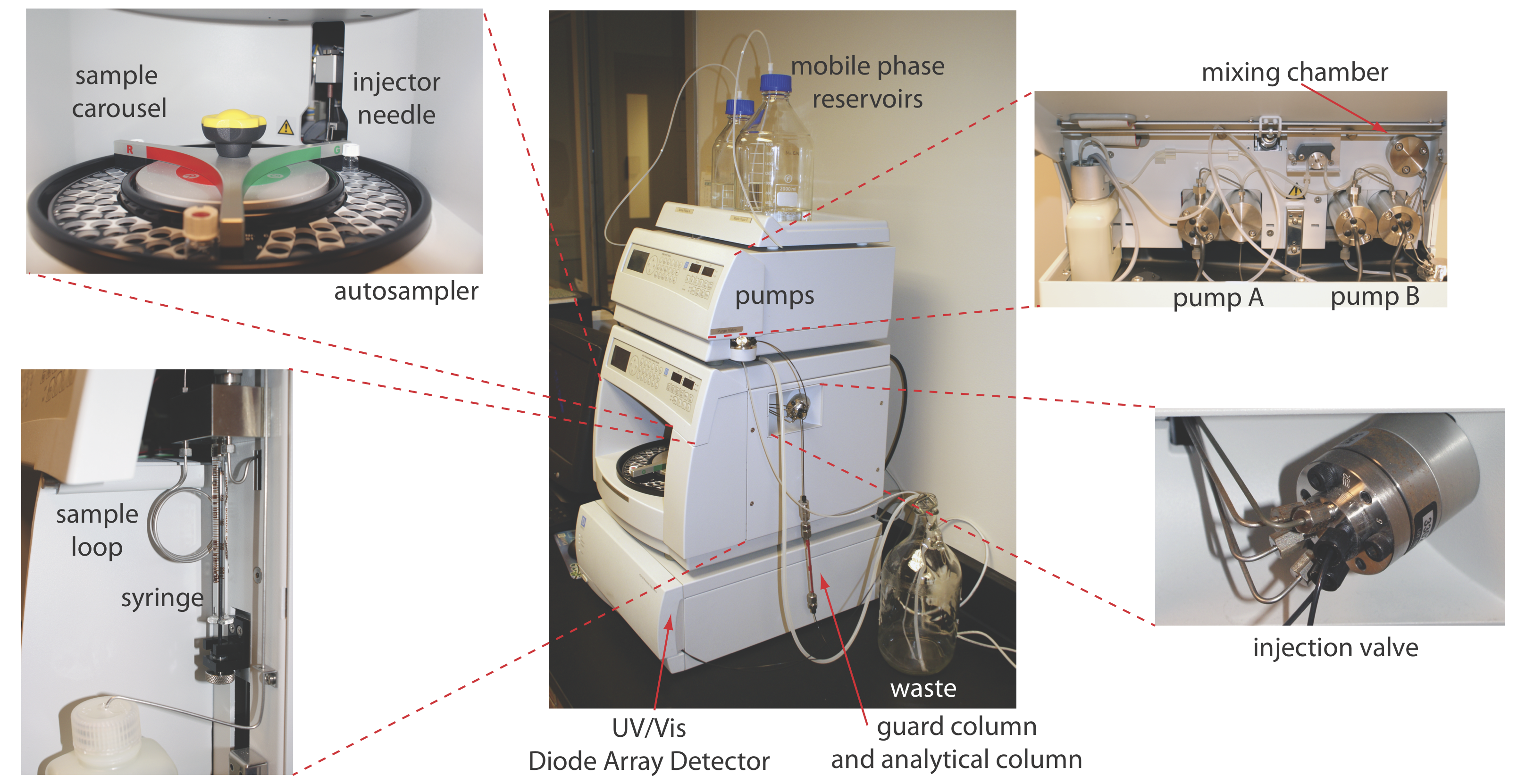
El tiempo de retención de soluto en HPLC se determina por su interacción con la fase estacionaria y la fase móvil. Hay varios tipos diferentes de interacciones soluto/fase estacionaria, incluyendo adsorción líquido-sólido, reparto líquido-líquido, intercambio iónico y exclusión por tamaño. Este capítulo trata exclusivamente de las separaciones por HPLC basadas en el reparto líquido-líquido. Otras formas de cromatografía líquida reciben consideración en el Capítulo 12.6.
Columnas de HPLC
Una HPLC normalmente incluye dos columnas: una columna analítica, que es responsable de la separación, y una columna de protección que se coloca antes de la columna analítica para protegerla de la contaminación.
Columnas Analíticas
El tipo más común de columna de HPLC es un tubo de acero inoxidable con un diámetro interno entre 2.1 mm y 4.6 mm y una longitud entre 30 mm y 300 mm (Figura 12.5.2 ). La columna está empaquetada con partículas porosas de sílice de 3—10 µm con forma irregular o esférica. Las eficiencias típicas de la columna son 40000—60000 placas teóricas/m. Suponiendo una V máx/V min de aproximadamente 50, una columna de 25 cm con 50 000 placas/m tiene 12 500 placas teóricas y una capacidad máxima de 110.

Las columnas capilares utilizan menos disolvente y, debido a que la muestra se diluye en menor medida, producen señales más grandes en el detector. Estas columnas están hechas de capilares de sílice fundida con diámetros internos de 44—200 μm y longitudes de 50—250 mm. Se han preparado columnas capilares empaquetadas con partículas de 3—5 μm con eficiencias de columna de hasta 250 000 placas teóricas [Novotony, M. Science, 1989, 246, 51—57].
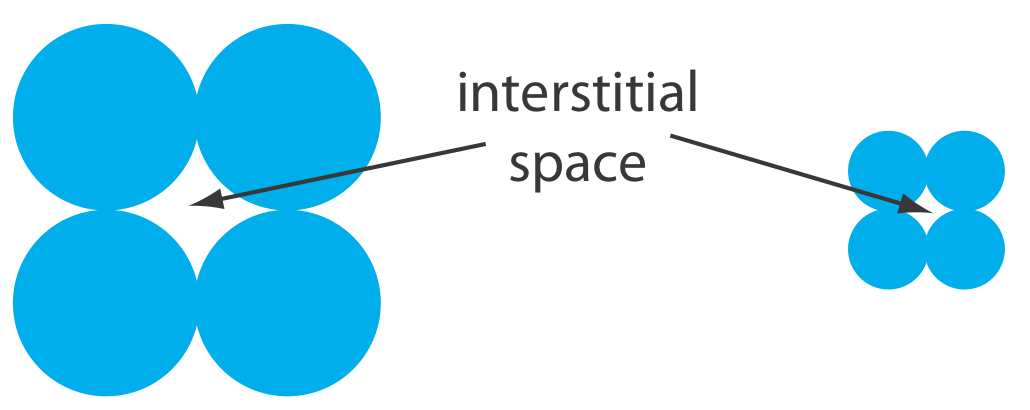
Una limitación para una columna capilar empaquetada es la contrapresión que se desarrolla al bombear la fase móvil a través de los pequeños espacios intersticiales entre el material de empaquetamiento de tamaño micrométrico particulado (Figura 12.5.3 ). Debido a que los tubos y accesorios que llevan la fase móvil tienen límites de presión, una contrapresión más alta requiere un caudal más bajo y un tiempo de análisis más largo. Las columnas monolíticas, en las que el soporte sólido es una sola varilla porosa, ofrecen eficiencias de columna equivalentes a una columna capilar empaquetada al tiempo que permiten caudales más rápidos. Una columna monolítica, que generalmente es similar en tamaño a una columna empaquetada convencional, aunque también hay disponibles columnas capilares más pequeñas, se prepara formando la varilla monolítica en un molde y cubriéndola con tubos de PTFE o una resina polimérica. Las varillas monolíticas hechas de un polímero de gel de sílice suelen tener macroporos con diámetros de aproximadamente 2 μm y mesoporos—poros dentro de los macróporos—con diámetros de aproximadamente 13 nm [Cabrera, K. Chromatography Online, 1 de abril de 2008].
Columnas de Guardia
Dos problemas tienden a acortar la vida útil de una columna analítica. Primero, los solutos que se unen irreversiblemente a la fase estacionaria degradan el rendimiento de la columna al disminuir la cantidad de fase estacionaria disponible para efectuar una separación. Segundo, el material particulado inyectado con la muestra puede obstruir la columna analítica. Para minimizar estos problemas colocamos una columna de guarda antes de la columna analítica. Una columna Guard generalmente contiene el mismo material de relleno particulado y fase estacionaria que la columna analítica, pero es significativamente más corta y menos cara, una longitud de 7.5 mm y un costo de una décima parte del de la columna analítica correspondiente es típica. Debido a que están destinados a ser sacrificiales, las columnas de guardia se reemplazan regularmente.
Si observa de cerca la Figura 12.5.1 , verá la pequeña columna de guarda justo encima de la columna analítica.
Fases estacionarias para cromatografía líquido-líquida
En la cromatografía líquido-líquido, la fase estacionaria es una película líquida recubierta sobre un material de empaque, típicamente partículas de sílice porosas de 3—10 μm. Debido a que la fase estacionaria puede ser parcialmente soluble en la fase móvil, puede eluirse o sangrar de la columna con el tiempo. Para evitar la pérdida de fase estacionaria, lo que acorta la vida útil de la columna, se une covalentemente a las partículas de sílice. Las fases estacionarias unidas se crean haciendo reaccionar las partículas de sílice con un organoclorosilano de la forma general Si (CH 3) 2 RCl, donde R es un grupo alquilo o alquilo sustituido.
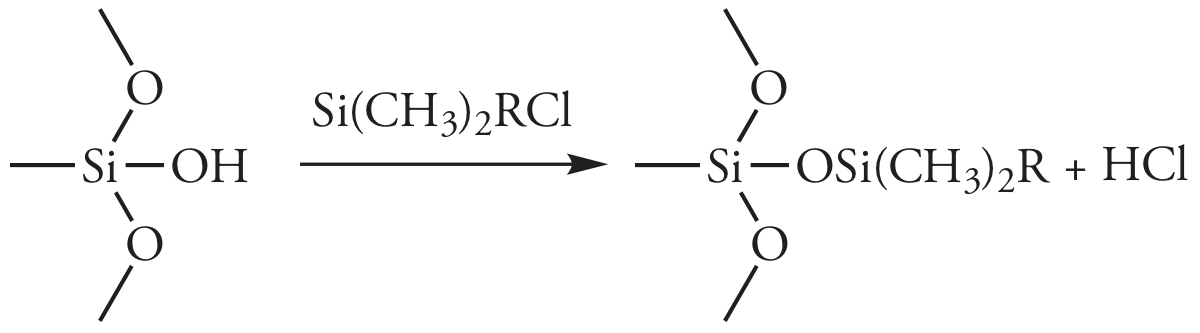
Para evitar interacciones no deseadas entre los solutos y cualquier grupo —SiOH restante, se usa Si (CH 3) 3 Cl para convertir sitios sin reaccionar en\(–\text{SiOSi(CH}_3)_3\); tales columnas se designan como terminalmente rematadas.
Las propiedades de una fase estacionaria dependen del grupo alquilo del organosilano. Si R es un grupo funcional polar, entonces la fase estacionaria es polar. Ejemplos de fases estacionarias polares incluyen aquellas en las que R contiene un grupo funcional ciano (—C 2 H 4 CN), un diol (—C 3 H 6 OCH 2 CHOHCH 2 OH) o un grupo funcional amino (—C 3 H 6 NH 2). Debido a que la fase estacionaria es polar, la fase móvil es un disolvente no polar o moderadamente polar. La combinación de una fase estacionaria polar y una fase móvil no polar se denomina cromatografía de fase normal.
En la cromatografía de fase inversa, que es la forma más común de HPLC, la fase estacionaria es no polar y la fase móvil es polar. Las fases estacionarias no polares más comunes utilizan un organoclorosilano donde el grupo R es una cadena hidrocarbonada n-octilo (C 8) o n-octildecilo (C 18). La mayoría de las separaciones de fase inversa se llevan a cabo usando una solución acuosa tamponada como fase móvil polar, o usando otros disolventes polares, como metanol y acetonitrilo. Debido a que el sustrato de sílice puede sufrir hidrólisis en soluciones básicas, el pH de la fase móvil debe ser inferior a 7.5.
Parece extraño que la forma más común de cromatografía líquida se identifique como fase inversa en lugar de fase normal. Tal vez recuerde que uno de los primeros ejemplos de cromatografía fue la separación de pigmentos vegetales por parte de Mikhail Tswett utilizando una columna polar de carbonato de calcio y una fase móvil no polar de éter de petróleo. La asignación de normal e invertida, por lo tanto, tiene que ver con la precedencia.
Fases Móviles
El orden de elución de los solutos en HPLC se rige por la polaridad. Para una separación de fase normal, un soluto de menor polaridad pasa proporcionalmente menos tiempo en la fase estacionaria polar y eluye ante un soluto que es más polar. Dada una fase estacionaria particular, los tiempos de retención en HPLC de fase normal se controlan ajustando las propiedades de la fase móvil. Por ejemplo, si la resolución entre dos solutos es pobre, el cambio a una fase móvil menos polar mantiene los solutos en la columna por más tiempo y brinda más oportunidades para su separación. En HPLC de fase inversa el orden de elución es el opuesto al de una separación de fase normal, con solutos más polares eluyendo primero. El aumento de la polaridad de la fase móvil conduce a tiempos de retención más largos. Los tiempos de retención más cortos requieren una fase móvil de menor polaridad.
Elección de una Fase Móvil: Uso del Índice de Polaridad
Existen varios índices que ayudan a seleccionar una fase móvil, uno de los cuales es el índice de polaridad [Snyder, L. R.; Glajch, J. L.; Kirkland, J. J. Practical HPLC Method Development, Wiley-Inter- science: New York, 1988]. El cuadro 12.5.1 proporciona valores del índice de polaridad\(P^{\prime}\), para varias fases móviles comunes, donde valores mayores de\(P^{\prime}\) corresponden a disolventes más polares. Mezclar dos o más fases móviles, asumiendo que son miscibles, crea una fase móvil de polaridad intermedia. Por ejemplo, una fase móvil binaria preparada combinando el disolvente A y el disolvente B tiene un índice de polaridad\(P_{AB}^{\prime}\), de
\[P_{A B}^{\prime}=\Phi_{A} P_{A}^{\prime}+\Phi_{B} P_{B}^{\prime} \label{12.1}\]
donde\(P_A^{\prime}\) y\(P_B^{\prime}\) son los índices de polaridad para los solventes A y B, y\(\Phi_A\) y\(\Phi_B\) son las fracciones de volumen para los dos solventes.
Se realiza una separación por HPLC de fase inversa utilizando una fase móvil de 60% v/v de agua y 40% v/v de metanol. ¿Cuál es el índice de polaridad de la fase móvil?
Solución
Usando la Ecuación\ ref {12.1} y los valores de la Tabla 12.5.1 , el índice de polaridad para una mezcla de 60:40 agua-metanol es
\[P_{A B}^{\prime}=\Phi_\text{water} P_\text{water}^{\prime}+\Phi_\text{methanol} P_\text{methanol}^{\prime} \nonumber\]
\[P_{A B}^{\prime}=0.60 \times 10.2+0.40 \times 5.1=8.2 \nonumber\]
Supongamos que necesita una fase móvil con un índice de polaridad de 7.5. Explica cómo puedes preparar esta fase móvil usando metanol y agua.
- Contestar
-
Si dejamos que x sea la fracción de agua en la fase móvil, entonces 1 — x es la fracción de metanol. Sustituyendo estos valores en la Ecuación\ ref {12.1} y resolviendo para x
\[7.5=10.2 x+5.1(1-x) \nonumber\]
\[7.5=10.2 x+5.1-5.1 x \nonumber\]
\[2.4=5.1 x \nonumber\]
da x como 0.47. La fase móvil es 47% v/v de agua y 53% v/v de metanol.
Como regla general, un cambio de dos unidades en el índice de polaridad corresponde a un cambio de aproximadamente 10 veces en el factor de retención de un soluto. Aquí hay un ejemplo sencillo. Si el factor de retención de un soluto, k, es 22 cuando se usa agua como fase móvil (\(P^{\prime}\)= 10.2), entonces cambiar a una fase móvil de 60:40 agua-metanol (\(P^{\prime}\)= 8.2) disminuye k a aproximadamente 2.2. Tenga en cuenta que el factor de retención se vuelve más pequeño porque estamos cambiando de una fase móvil más polar a una fase móvil menos polar en una separación de fase inversa.
Elección de una fase móvil: Ajuste de la selectividad
Cambiar el índice de polaridad de la fase móvil cambia el factor de retención de un soluto. Como aprendimos en el Capítulo 12.3, sin embargo, un cambio en k no es una manera efectiva de mejorar la resolución cuando el valor inicial de k es mayor que 10. Para efectuar una mejor separación entre dos solutos debemos mejorar el factor de selectividad,\(\alpha\). Existen dos métodos comunes para aumentar\(\alpha\): agregar un reactivo a la fase móvil que reacciona con los solutos en una reacción de equilibrio secundario o cambiar a una fase móvil diferente.
Aprovechar una reacción de equilibrio secundario es una estrategia útil para mejorar una separación [(a) Foley, J. P. Chromatography, 1987, 7, 118—128; (b) Foley, J. P.; May, W. E. Anal. Chem. 1987, 59, 102—109; (c) Foley, J. P.; May, W. E. Anal. Chem. 1987, 59, 110—115]. La Figura 12.3.3, que consideramos anteriormente en este capítulo, muestra la separación en fase inversa de cuatro ácidos débiles —ácido benzoico, ácido tereftálico, ácido p-aminbenzoico y ácido p-hidroxibenzoico — en una columna C 18 no polar utilizando un tampón acuoso de ácido acético y acetato de sodio como fase móvil. Los tiempos de retención para estos ácidos débiles son más cortos cuando se usa una fase móvil menos ácida porque cada soluto está presente en una forma de base débil aniónica que es menos soluble en la fase estacionaria no polar. Si el pH de la fase móvil es suficientemente ácido, los solutos están presentes como ácidos débiles neutros que son más solubles en la fase estacionaria y tardan más en eluirse. Debido a que los solutos ácidos débiles no tienen valores idénticos de p K a, el pH de la fase móvil tiene un efecto diferente en el tiempo de retención de cada soluto, lo que nos permite encontrar el pH óptimo para efectuar una separación completa de los cuatro solutos.
La química ácido-base no es el único ejemplo de una reacción de equilibrio secundario. Otros ejemplos incluyen el emparejamiento iónico, la complejación y la interacción de solutos con micelas. Consideraremos el último de estos en el Capítulo 12.7 cuando discutamos la cromatografía capilar electrocinética micelar.
En Ejemplo 12.5.1 aprendimos a ajustar la polaridad de la fase móvil mezclando dos disolventes. Un índice de polaridad, sin embargo, es solo una guía, y las mezclas binarias de fase móvil con índices de polaridad idénticos pueden no resolver igualmente un par de solutos. La Tabla 12.5.2 , por ejemplo, muestra tiempos de retención para cuatro ácidos débiles en dos fases móviles con valores casi idénticos para\(P^{\prime}\). Aunque el orden de elución es el mismo para ambas fases móviles, el tiempo de retención de cada soluto se ve afectado de manera diferente por la elección del disolvente orgánico. Si cambiamos de usar acetonitrilo a tetrahidrofurano, por ejemplo, encontramos que el ácido benzoico eluye más rápidamente y que el ácido p-hidroxibenzoico eluye más lentamente. Aunque podemos resolver completamente estos dos solutos usando una fase móvil que es 16% v/v acetonitrilo, no podemos resolverlos si la fase móvil es 10% tetrahidrofurano.
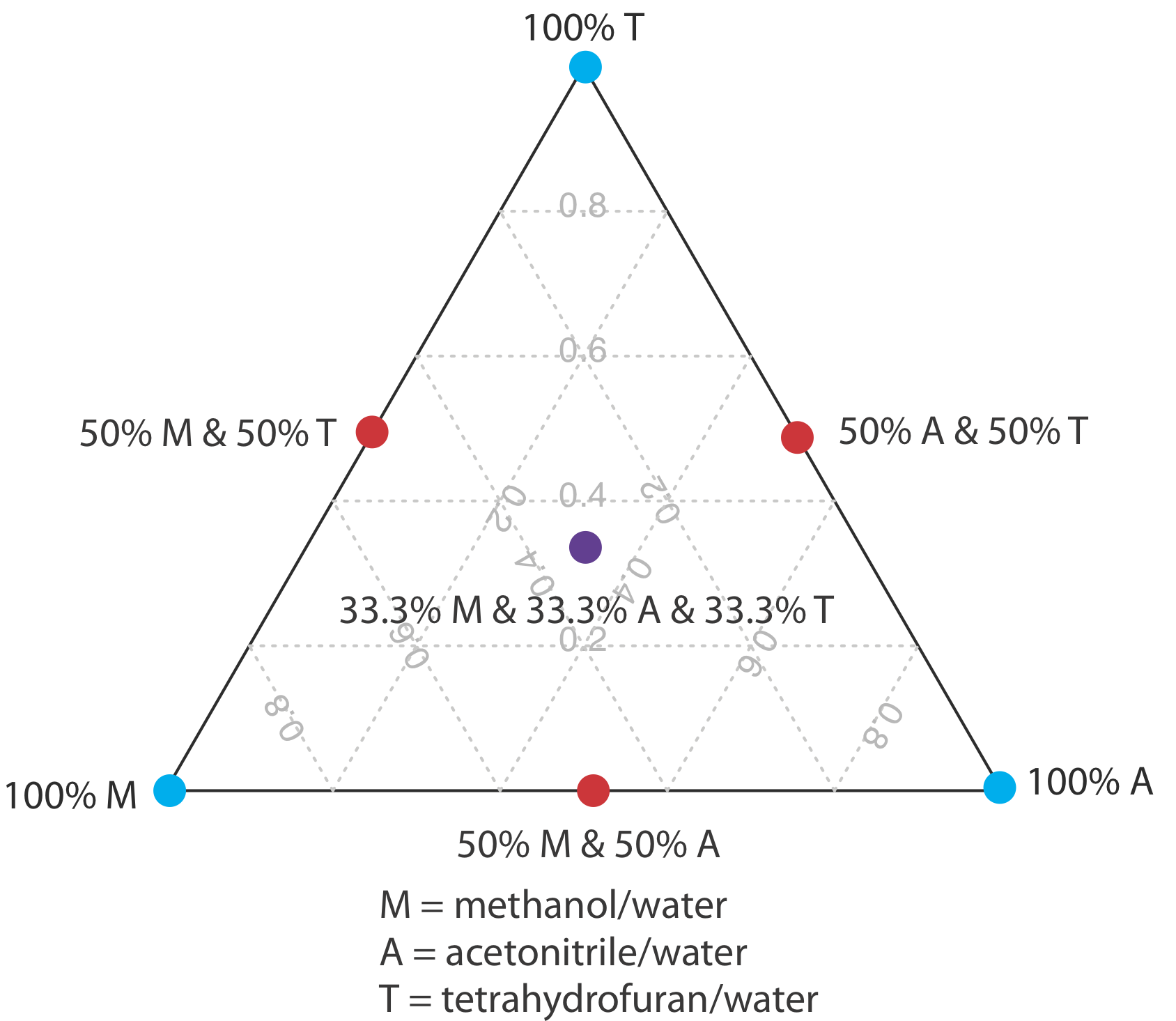
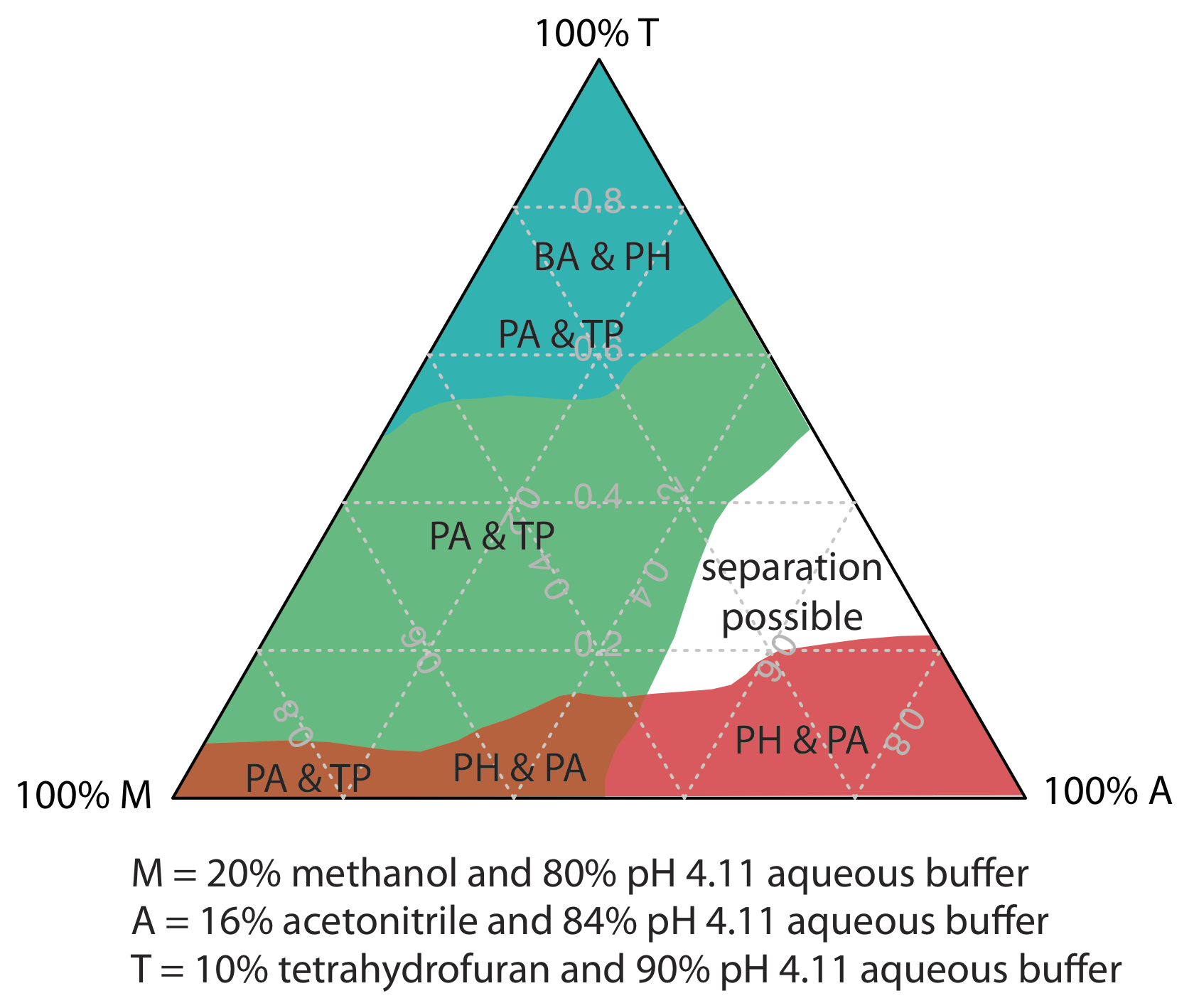
Una estrategia para encontrar la mejor fase móvil es utilizar el triángulo solvente mostrado en la Figura 12.5.4 , lo que nos permite explorar una amplia gama de fases móviles con solo siete experimentos. Comenzamos ajustando la cantidad de acetonitrilo en la fase móvil para producir la mejor separación posible dentro del tiempo de análisis deseado. A continuación, utilizamos la Tabla 12.5.3 para estimar la composición de las fases móviles de metanol/h 2 O y tetrahidrofurano/h 2 O que producirán tiempos de análisis similares. Se preparan cuatro fases móviles adicionales utilizando las fases móviles binarias y ternarias mostradas en la Figura 12.5.4 . Cuando examinamos los cromatogramas de estas siete fases móviles podemos encontrar que una o más proporcionan una separación adecuada, o podemos identificar una región dentro del triángulo solvente donde una separación es factible. La Figura 12.5.5 muestra un mapa de resolución para la separación en fase inversa de ácido benzoico, ácido tereftálico, ácido p-aminbenzoico y ácido p-hidroxibenzoico en una columna C 18 no polar en la que el tiempo máximo de análisis deseado se establece en 6 min [Harvey, D. T.; Byerly, S.; Bowman, A.; Tomlin, J. J. Chem. Educ. 1991, 68, 162—168]. Las áreas en azul, verde y rojo muestran composiciones de fase móvil que no proporcionan resolución basal. El área no sombreada representa composiciones de fase móvil donde es posible una separación.
La elección para comenzar con acetonitrilo es arbitraria; podemos elegir fácilmente comenzar con metanol o tetrahidrofurano.
Elección de una Fase Móvil: Eluciones Isocráticas y Gradientes
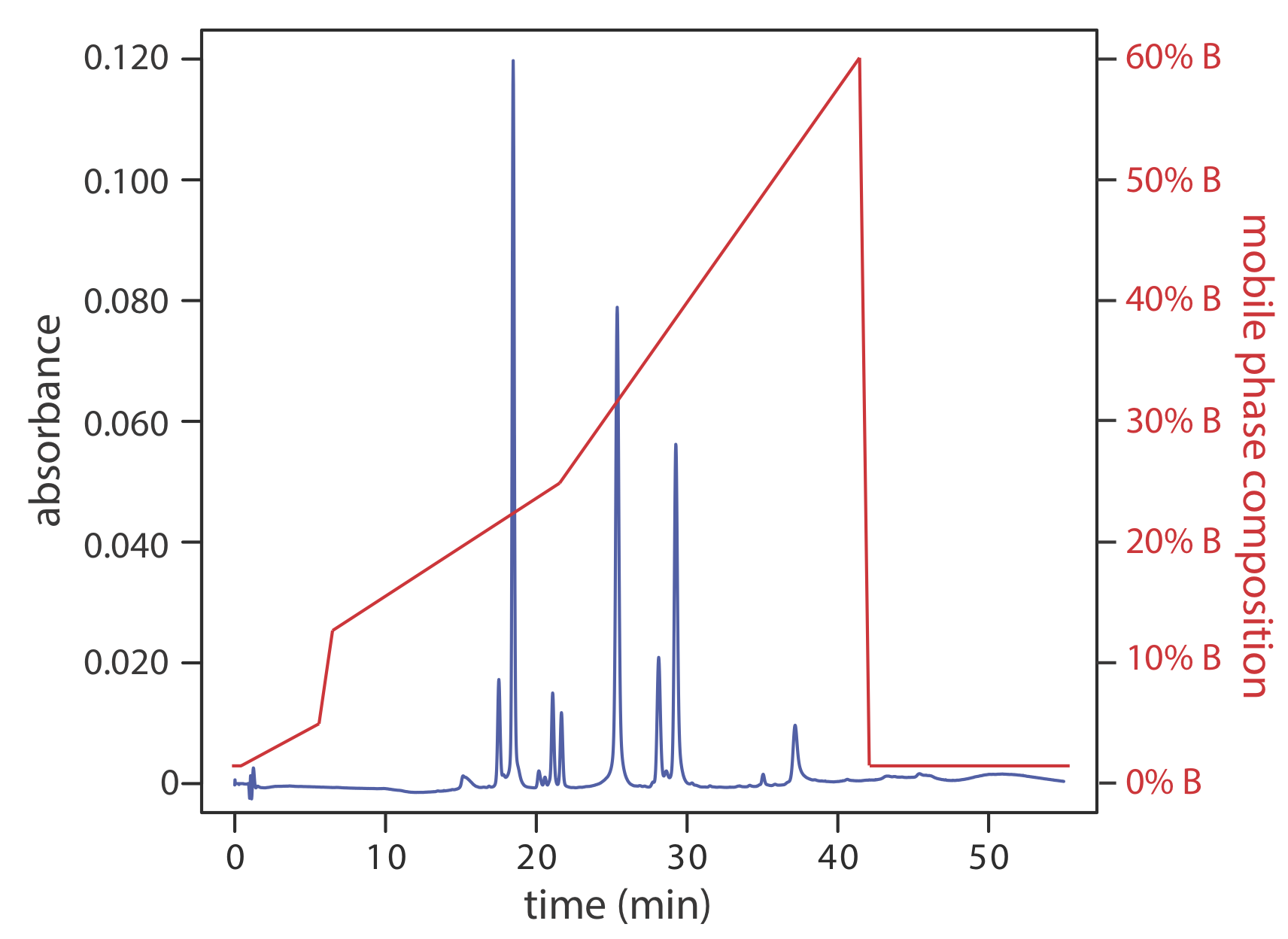
Una separación usando una fase móvil que tiene una composición fija es una elución isocrática. Una dificultad con una elución isocrática es que una fuerza de fase móvil apropiada para resolver solutos de elución temprana puede conducir a tiempos de retención inaceptablemente largos para solutos de elución tardía. La optimización de la fase móvil para solutos de elución tardía, por otro lado, puede proporcionar una separación inadecuada de solutos de elución temprana. Cambiar la composición de la fase móvil a medida que avanza la separación es una solución a este problema. Para una separación de fase inversa utilizamos una fase móvil inicial que es más polar. A medida que avanza la separación, ajustamos la composición de la fase móvil para que se vuelva menos polar (ver Figura 12.5.6 ). Tales separaciones se denominan eluciones de gradiente.
Plomería HPLC
En un cromatógrafo de gases la presión de un cilindro de gas comprimido es suficiente para empujar la fase móvil a través de la columna. Empujar una fase móvil líquida a través de una columna, sin embargo, requiere mucho más esfuerzo, generando presiones superiores a varios cientos de atmósferas. En esta sección consideramos la plomería básica necesaria para mover la fase móvil a través de la columna e inyectar la muestra a la fase móvil.
Mover la fase móvil
Una HPLC típica incluye entre 1 y 4 depósitos para almacenar disolventes de fase móvil. El instrumento en la Figura 12.5.1 , por ejemplo, tiene dos depósitos de fase móvil que se utilizan para una elución isocrática o una elución en gradiente mediante la extracción de disolventes de uno o ambos depósitos.
Antes de usar un disolvente de fase móvil debemos eliminar los gases disueltos, como N 2 y O 2, y pequeñas partículas, como el polvo. Debido a que hay una gran caída de presión a través de la columna, la presión en la entrada de la columna es de varios cientos de atmósferas, pero es la presión atmosférica a la salida de la columna, los gases disueltos en la fase móvil se liberan como burbujas de gas que pueden interferir con la respuesta del detector. La desgasificación se realiza de varias maneras, pero las más comunes son el uso de una bomba de vacío o el rociado con un gas inerte, como el He, que tiene una baja solubilidad en la fase móvil. Los materiales particulados, que pueden obstruir el tubo o columna de HPLC, se eliminan filtrando los disolventes.
El burbujeo de un gas inerte a través de la fase móvil libera gases volátiles disueltos. Este proceso se llama sparging.
Los disolventes de fase móvil se extraen de sus depósitos por la acción de una o más bombas. La Figura 12.5.7 muestra una vista en primer plano de las bombas para el instrumento en la Figura 12.5.1 . La bomba de trabajo y la bomba equilibradora tienen cada una un pistón cuyo movimiento de ida y vuelta mantiene un caudal constante de hasta varios ml/min y proporciona la alta presión de salida necesaria para empujar la fase móvil a través de la columna cromatográfica. En este instrumento en particular, cada bomba envía su fase móvil a una cámara de mezcla donde se combinan para formar la fase móvil final. La velocidad relativa de las dos bombas determina la composición final de la fase móvil.
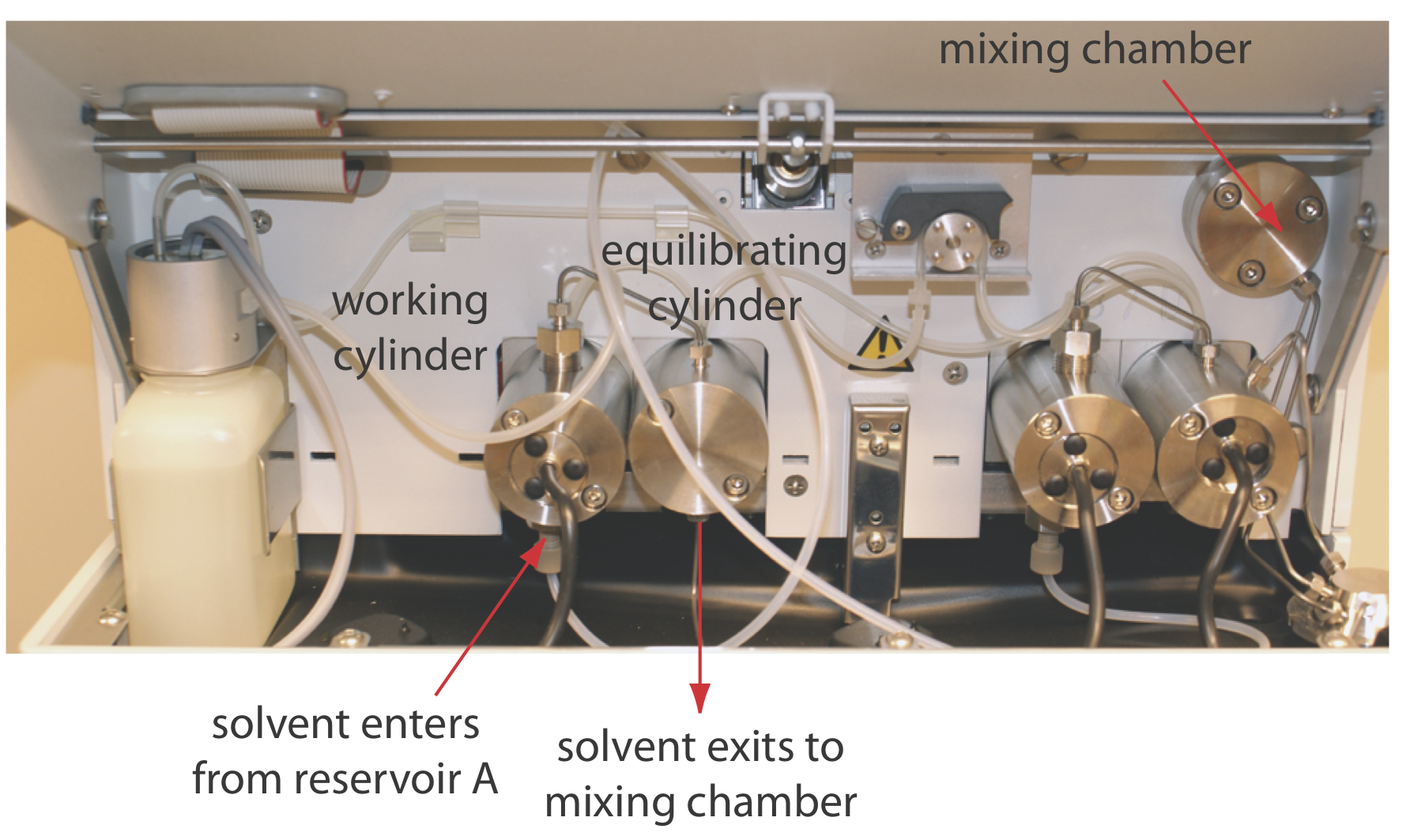
El movimiento de ida y vuelta de una bomba de vaivén crea un flujo pulsado que aporta ruido al cromatograma. Para minimizar estos pulsos, cada bomba de la Figura 12.5.7 tiene dos cilindros. Durante el movimiento delantero del cilindro de trabajo, llena el cilindro equilibrador y establece el flujo a través de la columna. Cuando el cilindro de trabajo está en su carrera inversa, el flujo es mantenido por el pistón en el cilindro de equilibrio. El resultado es un flujo libre de pulsos.
Existen otras formas posibles de controlar la composición y el caudal de la fase móvil. Por ejemplo, en lugar de las dos bombas de la Figura 12.5.7 , podemos colocar una válvula dosificadora de disolvente antes de una sola bomba. El valor de dosificación de solvente conecta dos o más depósitos de solvente a la bomba y determina cuánto de cada solvente se extrae durante cada uno de los ciclos de la bomba. Otro enfoque para eliminar un flujo pulsado es incluir un amortiguador de pulsos entre la bomba y la columna. Un amortiguador de pulsos es una cámara llena de un fluido fácilmente comprimido y un diafragma flexible. Durante la carrera hacia adelante del pistón, el fluido en el amortiguador de impulsos se comprime. Cuando el pistón se retira para rellenar la bomba, la presión del fluido de expansión en el amortiguador de pulsos mantiene el caudal.
Inyección de la muestra
La presión de operación dentro de una HPLC es suficientemente alta como para que no podamos inyectar la muestra en la fase móvil insertando una jeringa a través de un septo, como es posible en la cromatografía de gases. En su lugar, inyectamos la muestra usando un inyector de bucle, cuyo diagrama se muestra en la Figura 12.5.8 . En la posición de carga, un bucle de muestra, que está disponible en una variedad de tamaños que van desde 0.5 μL hasta 5 ml, se aísla de la fase móvil y se abre a la atmósfera. El asa de muestra se llena usando una jeringa con una capacidad varias veces mayor que la del asa de muestra, con exceso de muestra saliendo por la línea de desechos. Después de cargar la muestra, el inyector se gira a la posición de inyección, que redirige la fase móvil a través del bucle de muestra y hacia la columna.
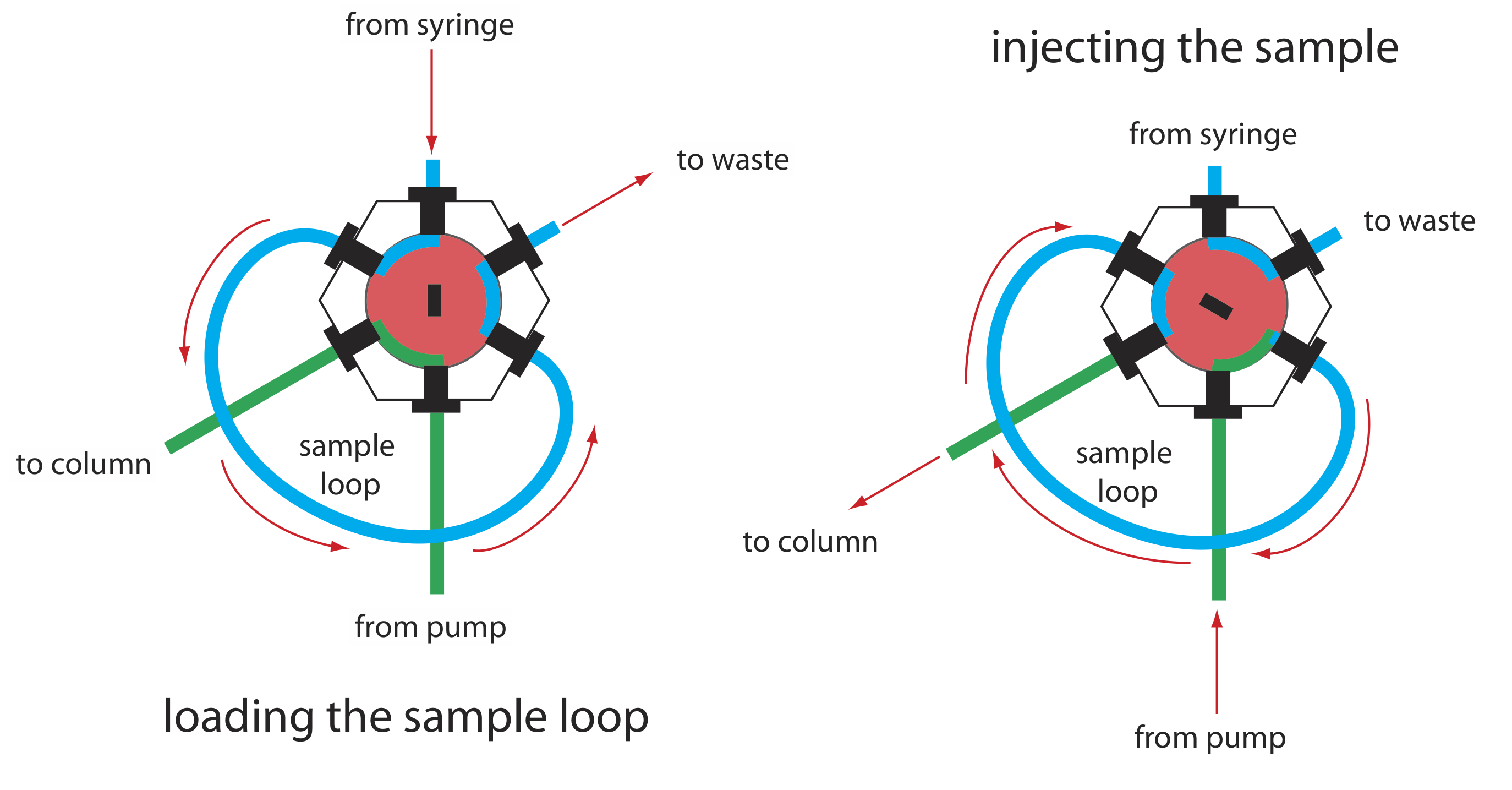
El instrumento en la Figura 12.5.1 utiliza un muestreador automático para inyectar muestras. En lugar de usar una jeringa para empujar la muestra hacia el bucle de muestra, la jeringa extrae la muestra hacia el bucle de muestra.
Detectores para HPLC
Se han utilizado muchos tipos diferentes de detectores para monitorear las separaciones por HPLC, la mayoría de los cuales utilizan las técnicas espectroscópicas del Capítulo 10 o las técnicas electroquímicas del Capítulo 11.
Detectores espectroscópicos
Los detectores de HPLC más populares aprovechan el espectro de absorción UV/Vis de un analito. Estos detectores van desde diseños simples, en los que la longitud de onda analítica se selecciona utilizando filtros apropiados, hasta un espectrofotómetro modificado en el que el compartimento de muestra incluye una celda de flujo. La Figura 12.5.9 muestra el diseño de una celda de flujo típica cuando se usa un espectrómetro de matriz de diodos como detector. La celda de flujo tiene un volumen de 1—10 μL y una longitud de trayectoria de 0.2—1 cm.
Para revisar los detalles de cómo medimos la absorbancia, consulte el Capítulo 10.2. Más información sobre diferentes tipos de instrumentos, incluido el espectrómetro de matriz de diodos, se encuentra en el Capítulo 10.3.
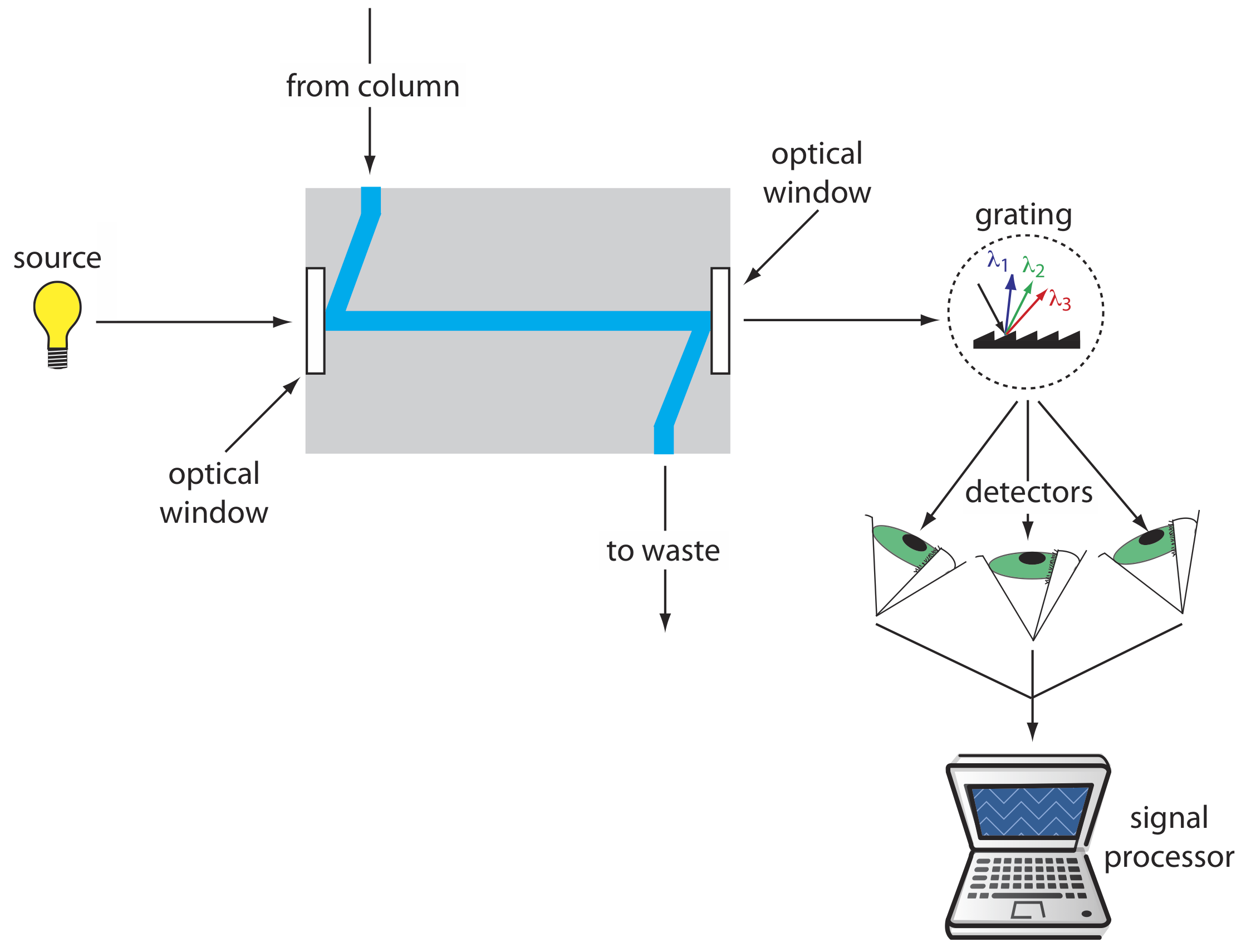
Cuando se usa un detector UV/Vis el cromatograma resultante es una gráfica de absorbancia en función del tiempo de elución (ver Figura 12.5.10 ). Si el detector es un espectrómetro de matriz de diodos, entonces también podemos mostrar el resultado como un cromatograma tridimensional que muestra la absorbancia en función de la longitud de onda y el tiempo de elución. Una limitación al uso de la absorbancia es que la fase móvil no puede absorber en las longitudes de onda que deseamos monitorear. La Tabla 12.5.1 enumera la longitud de onda UV mínima útil para varios solventes de HPLC comunes. Los detectores de absorbancia proporcionan límites de detección de tan solo 100 pg-1 ng de analito inyectado.
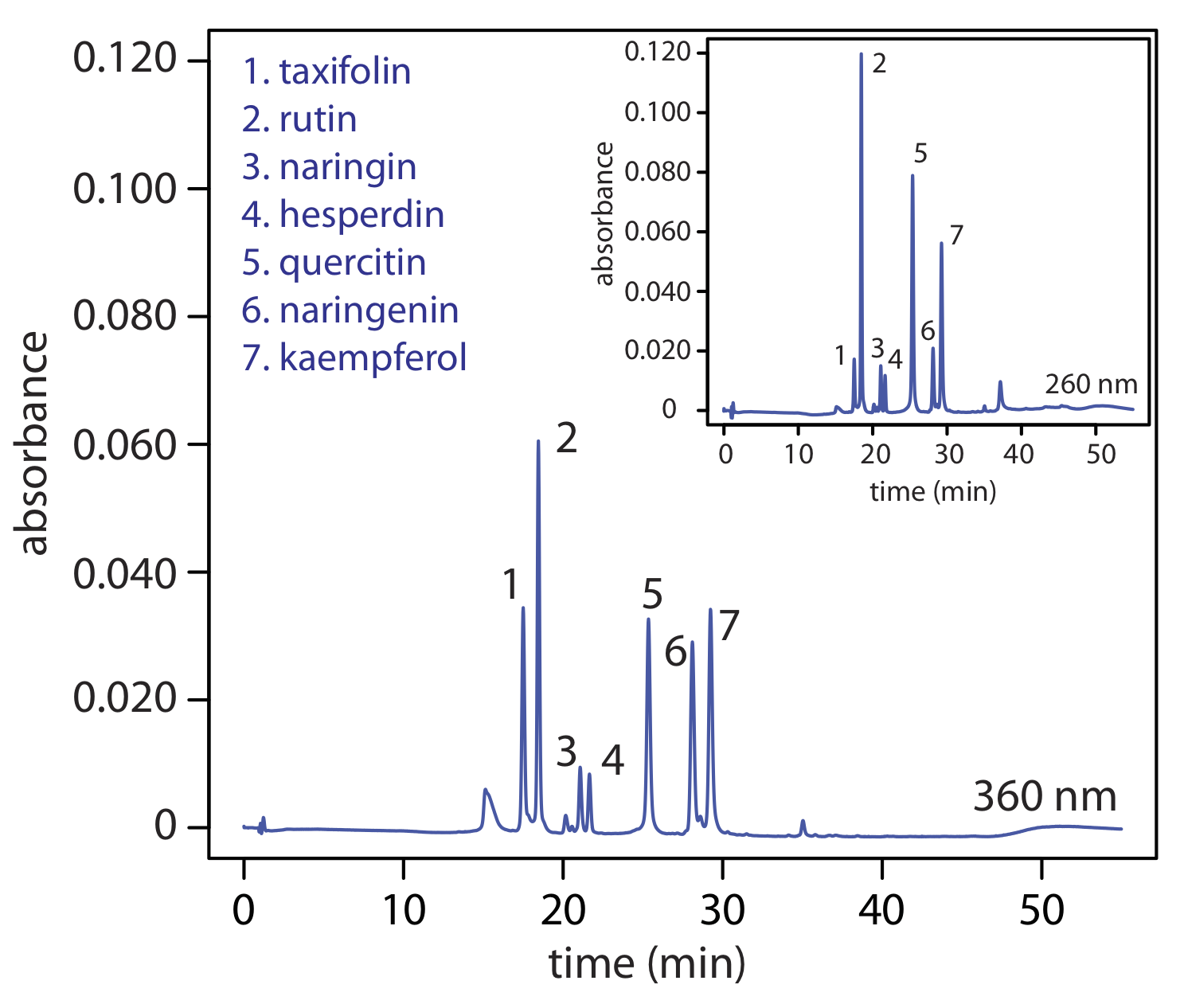
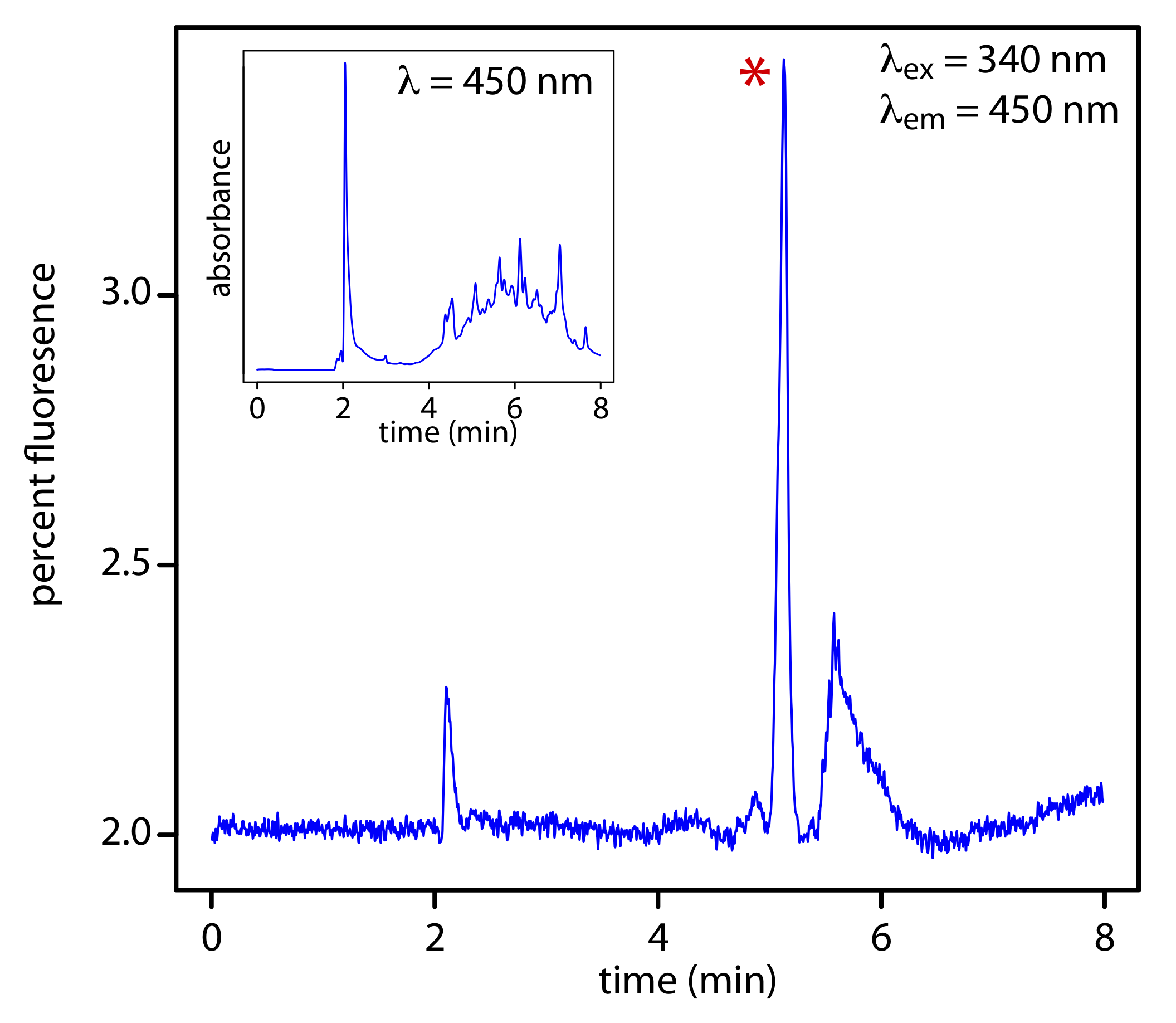
Si un analito es fluorescente, podemos colocar la celda de flujo en un espectrofluorímetro. Como se muestra en la Figura 12.5.11 , un detector de fluorescencia proporciona selectividad adicional porque solo algunos de los componentes de una muestra son fluorescentes. Los límites de detección son tan pequeños como 1—10 pg de analito inyectado.
Consulte el Capítulo 10.6 para una revisión de espectroscopía de fluorescencia y espectrofluorímetros.
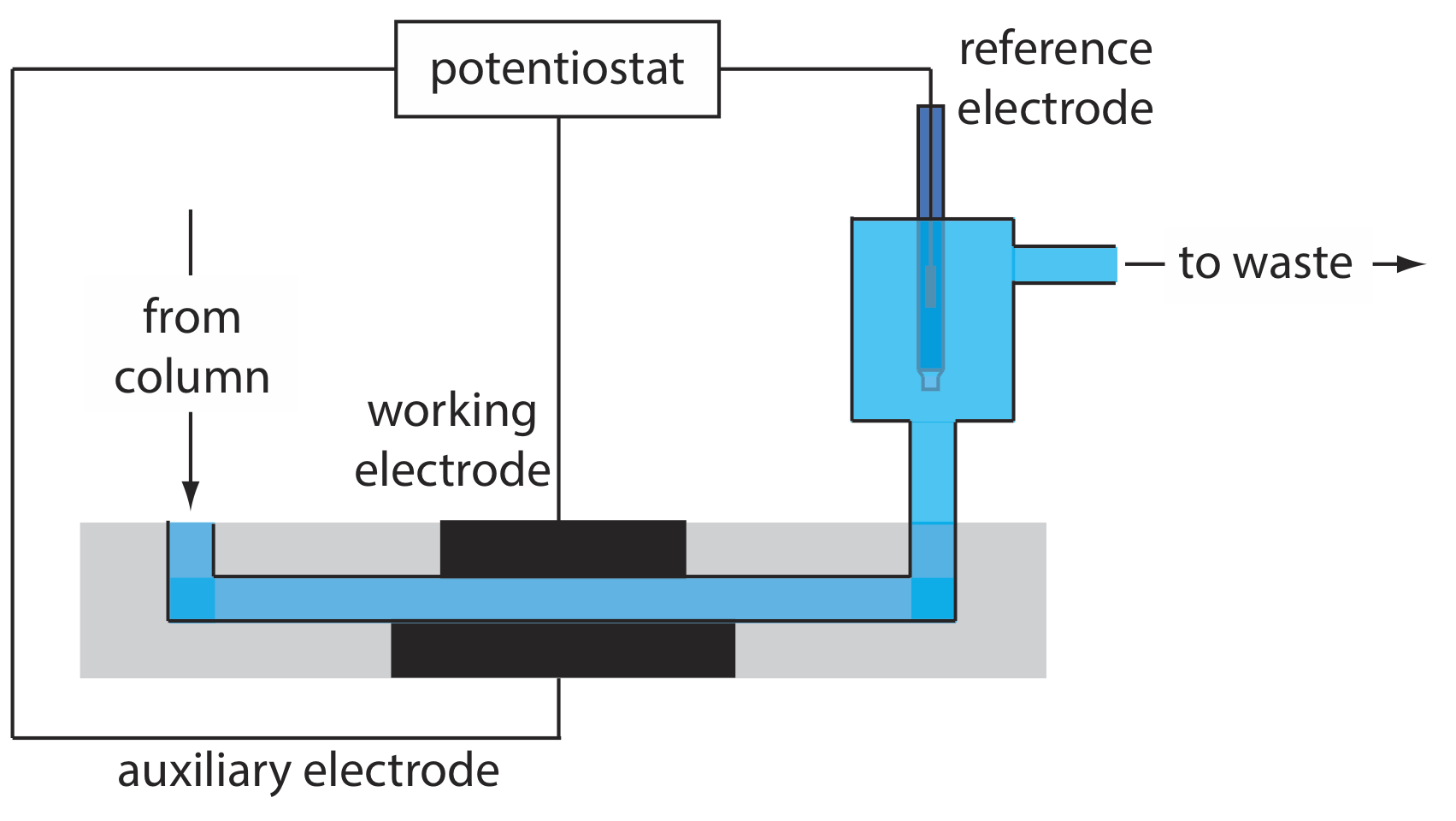
Detectores Electroquímicos
Otro grupo común de detectores de HPLC son aquellos basados en mediciones electroquímicas como amperometría, voltamperometría, culombimetría y conductividad. La figura 12.5.12 , por ejemplo, muestra una celda de flujo amperométrico. El efluente de la columna pasa sobre el electrodo de trabajo, mantenido a un potencial constante en relación con un electrodo de referencia aguas abajo, que oxida o reduce completamente los analitos. La corriente que fluye entre el electrodo de trabajo y el electrodo auxiliar sirve como señal analítica. Los límites de detección para la detección electroquímica amperométrica son de 10 pg-1 ng de analito inyectado.
Consulte el Capítulo 11.4 para una revisión de la amperometría.
Otros Detectores
Varios otros detectores han sido utilizados en HPLC. Medir un cambio en el índice de refracción de la fase móvil es análogo a monitorear la conductividad térmica de la fase móvil en cromatografía de gases. Un detector de índice de refracción es casi universal, respondiendo a casi todos los compuestos, pero tiene un límite de detección relativamente pobre de 0.1—1 μg de analito inyectado. Una limitación adicional de un detector de índice de refracción es que no puede usarse para una elución en gradiente a menos que los componentes de la fase móvil tengan índices de refracción idénticos.
Otro detector útil es un espectrómetro de masas. La Figura 12.5.13 muestra un diagrama de bloques de un instrumento HPLC—MS típico. El efluente de la columna ingresa a la fuente de iones del espectrómetro de masas utilizando una interfaz que elimina la mayor parte de la fase móvil, una necesidad esencial debido a la incompatibilidad entre la fase móvil líquida y el ambiente de alto vacío del espectrómetro de masas. En la cámara de ionización las moléculas restantes, una mezcla de los componentes de la fase móvil y los solutos, experimentan ionización y fragmentación. El analizador de masas del espectrómetro de masas separa los iones por su relación masa/carga (m/z). Un detector cuenta los iones y muestra el espectro de masas.
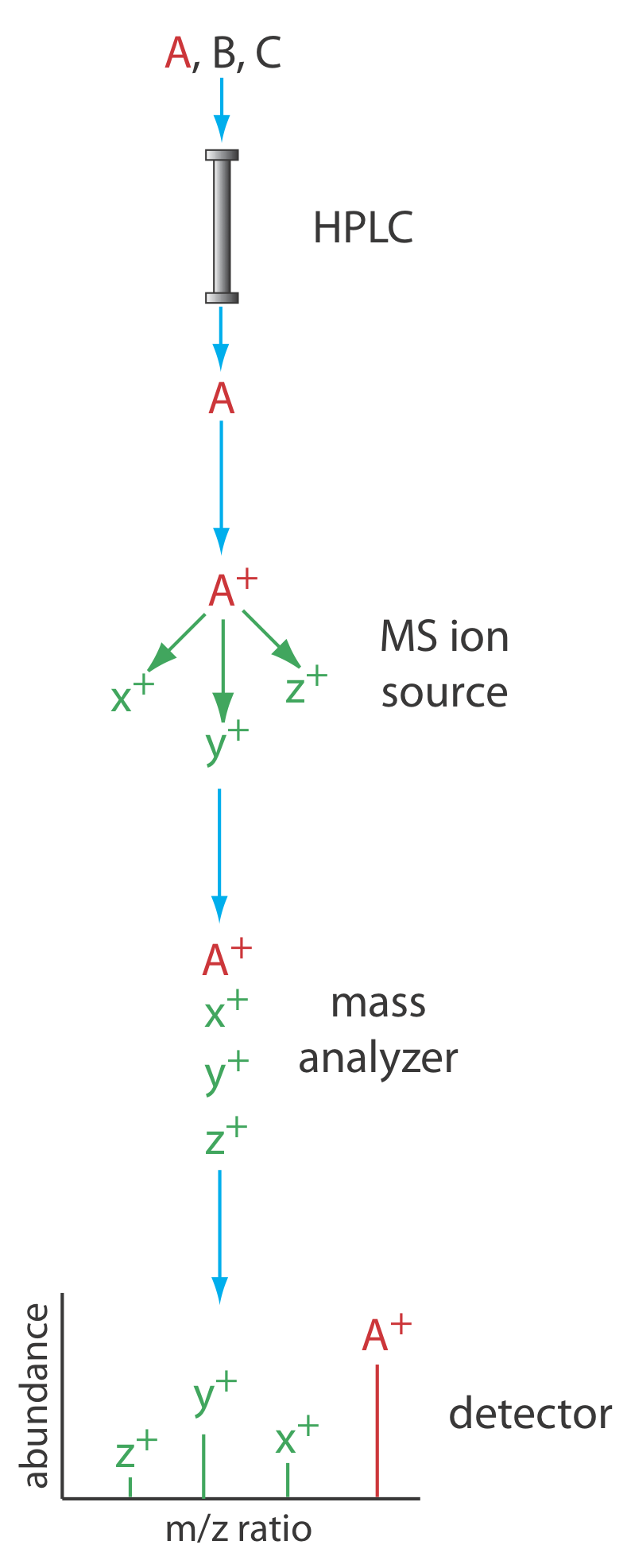
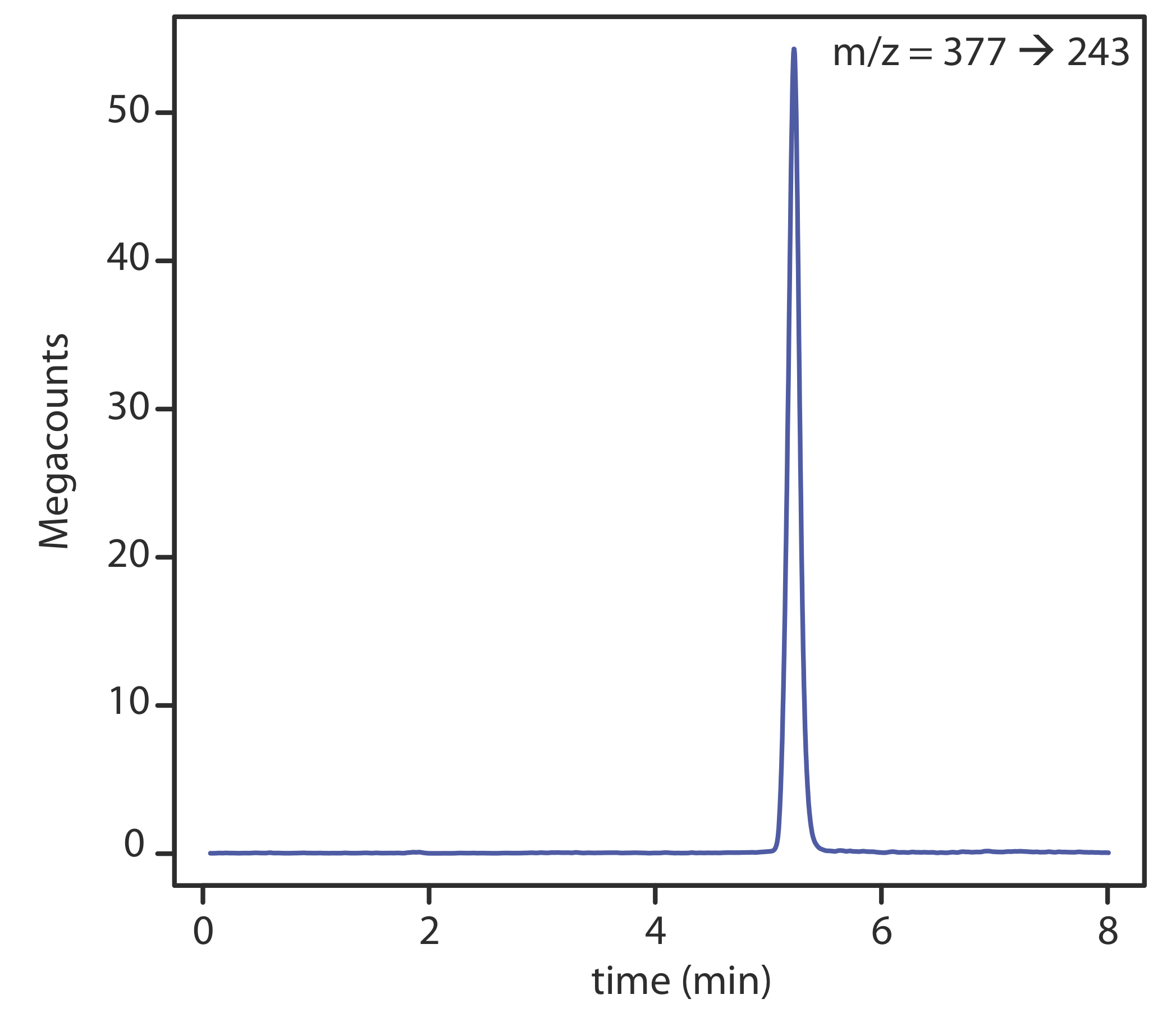
Existen varias opciones para monitorear el cromatograma cuando se usa un espectrómetro de masas como detector. El método más común es escanear continuamente todo el espectro de masas y reportar la señal total para todos los iones que llegan al detector durante cada exploración. Este escaneo de iones totales proporciona detección universal para todos los analitos. Como se ve en la Figura 12.5.14 , podemos lograr cierto grado de selectividad monitoreando solo relaciones masa-carga específicas, un proceso llamado monitoreo de iones selectivos.
Las ventajas de usar un espectrómetro de masas en HPLC son las mismas que para la cromatografía de gases. Los límites de detección son muy buenos, típicamente 0.1—1 ng de analito inyectado, con valores tan bajos como 1—10 pg para algunas muestras. Además, un espectrómetro de masas proporciona información cualitativa y estructural que puede ayudar a identificar los analitos. La interfaz entre la HPLC y el espectrómetro de masas es técnicamente más difícil que en un GC-MS debido a la incompatibilidad de una fase móvil líquida con el requerimiento de alto vacío del espectrómetro de masas.
Para más detalles sobre espectrometría de masas consulte Introducción a la espectrometría de masas de Michael Samide y Olujide Akinbo, un recurso que forma parte de la Biblioteca Digital de Ciencias Analíticas.
Aplicaciones Cuantitativas
La cromatografía líquida de alto rendimiento se utiliza rutinariamente para análisis cualitativos y cuantitativos de muestras ambientales, farmacéuticas, industriales, forenses, clínicas y de productos de consumo.
Preparación de muestras para el análisis
Las muestras en forma líquida se inyectan en la HPLC después de una limpieza adecuada para eliminar cualquier material particulado, o después de una extracción adecuada para eliminar los interferentes de la matriz. En la determinación de hidrocarburos poliaromáticos (PAH) en aguas residuales, por ejemplo, una extracción con CH 2 Cl 2 sirve para el doble propósito de concentrar los analitos y aislarlos de los interferentes de la matriz. Las muestras sólidas se disuelven primero en un disolvente adecuado o los analitos de interés se ponen en solución por extracción. Por ejemplo, un análisis de HPLC para los ingredientes activos y los productos de degradación en un comprimido farmacéutico a menudo comienza extrayendo el comprimido en polvo con una porción de fase móvil. Las muestras de gas se recolectan burbujeándolas a través de una trampa que contiene un solvente adecuado. Los isocianatos orgánicos en atmósferas industriales se recolectan burbujeando el aire a través de una solución de 1- (2-metoxifenil) piperazina en tolueno. La reacción entre los isocianatos y la 1- (2-metoxifenil) piperazina los estabiliza frente a la degradación antes del análisis por HPLC y los convierte en una forma química que puede ser monitoreada por absorción UV.
Cálculos cuantitativos
Un análisis por HPLC cuantitativo suele ser más fácil que un análisis de GC cuantitativo porque un bucle de muestra de volumen fijo proporciona una inyección más precisa y precisa. Como resultado, la mayoría de los métodos de HPLC cuantitativa no necesitan un estándar interno y, en su lugar, utilizan estándares externos y una curva de calibración normal.
Un estándar interno es necesario cuando se usa HPLC-MS porque la interfaz entre la HPLC y el espectrómetro de masas no permite una transferencia reproducible del eluyente de la columna a la cámara de ionización de la EM.
La concentración de hidrocarburos aromáticos polinucleares (PAH) en el suelo se determina extrayendo primero los HAP con cloruro de metileno. El extracto se diluye, si es necesario, y los HAP se separan por HPLC usando un detector de UV/Vis o fluorescencia. La calibración se logra usando uno o más estándares externos. En un análisis típico se extrae una muestra de 2.013 g de suelo seco con 20.00 mL de cloruro de metileno. Después de filtrar para eliminar la tierra, se retira una porción de 1.00-mL del extracto y se diluye a 10.00 mL con acetonitrilo. Inyectar 5 μL del extracto diluido en una HPLC da una señal de 0.217 (unidades arbitrarias) para el fluoranteno PAH. Cuando se analizan 5 μL de un estándar de fluoranteno de 20.0-ppm usando las mismas condiciones, se mide una señal de 0.258. Reportan las partes por millón de fluoranteno en el suelo.
Solución
Para un estándar externo de punto único, la relación entre la señal, S, y la concentración, C, de fluoranteno es
\[S = kC \nonumber\]
Sustituyendo en valores la señal y concentración del estándar da el valor de k como
\[k=\frac{S}{C}=\frac{0.258}{20.0 \text{ ppm}}=0.0129 \text{ ppm}^{-1} \nonumber\]
El uso de este valor para k y la señal de HPLC de la muestra da una concentración de fluoranteno de
\[C=\frac{S}{k}=\frac{0.217}{0.0129 \text{ ppm}^{-1}}=16.8 \text{ ppm} \nonumber\]
para la muestra de suelo extraída y diluida. La concentración de fluoranteno en el suelo es
\[\frac{16.8 \text{ g} / \mathrm{mL} \times \frac{10.00 \text{ mL}}{1.00 \text{ mL}} \times 20.00 \text{ mL}}{2.013 \text{ g} \text { sample }}=1670 \text{ ppm} \text { fluoranthene } \nonumber\]
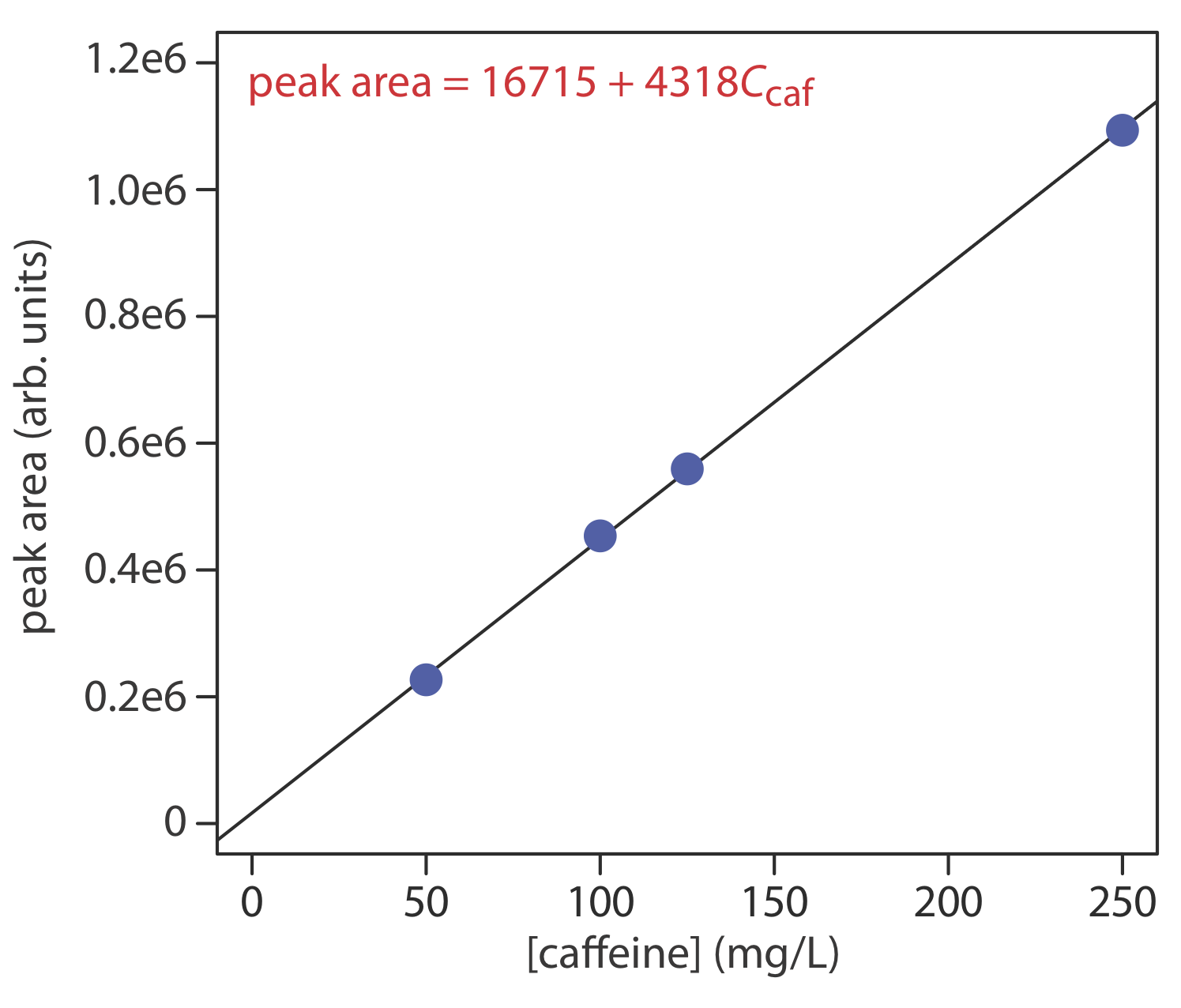
La concentración de cafeína en las bebidas se determina mediante una separación HPLC de fase inversa usando una fase móvil de 20% acetonitrilo y 80% agua, y usando una columna C 8 no polar. Los resultados de una serie de inyecciones de 10-μL de estándares de cafeína se encuentran en la siguiente tabla.
| [cafeína] (mg/L) | área de pico (unidades arb.) |
|---|---|
| 50.0 | 226724 |
| 100.0 | 453762 |
| 125.0 | 559443 |
| 250.0 | 1093637 |
¿Cuál es la concentración de cafeína en una muestra si una inyección de 10 μL da un área pico de 424195? Los datos en este problema provienen de Kusch, P.; Knupp, G. “Determinación Simultánea de Cafeína en Bebidas Cola y Otras Bebidas por HPTLC de Fase Inversa y HPLC de Fase Inversa”, Chem. Educador, 2003, 8, 201—205.
- Contestar
-
La siguiente figura muestra la curva de calibración y la ecuación de calibración para el conjunto de estándares externos. Al sustituir el área del pico de la muestra en la ecuación de calibración se obtiene la concentración de cafeína en la muestra como 94.4 mg/L.
La mejor manera de apreciar los detalles teóricos y prácticos discutidos en esta sección es examinar cuidadosamente un método analítico típico. Aunque cada método es único, la siguiente descripción de la determinación de fluoxetina en suero proporciona un ejemplo instructivo de un procedimiento típico. La descripción aquí se basa en Smyth, W. F. Analytical Chemistry of Complex Matricies, Wiley Teubner: Chichester, Inglaterra, 1996, pp. 187—189.
Método Representativo 12.5.1: Determinación de Fluoxetina en Suero
Descripción del método
Fluoxetina es otro nombre para el medicamento antidepresivo Prozac. La determinación de fluoxetina en suero es una parte importante del monitoreo de su uso terapéutico. El análisis se complica por la compleja matriz de muestras de suero. Una extracción en fase sólida seguida de un análisis por HPLC usando un detector de fluorescencia proporciona la selectividad y los límites de detección necesarios.
Procedimiento
Agregar una cantidad conocida del antidepresivo protriptilina, que sirve como patrón interno, a cada muestra de suero y a cada estándar externo. Para eliminar los interferentes de la matriz, pase una alícuota de 0.5 mL de cada muestra de suero o estándar a través de un cartucho de extracción en fase sólida C 18. Después de lavar el cartucho para eliminar los interferentes, eluir los constituyentes restantes, incluyendo el analito y el patrón interno, lavando el cartucho con 0.25 mL de una mezcla 25:75 v/v de HClo 4 0.1 M y acetonitrilo. Inyecte una alícuota de 20 μL en una columna de 15 cm y\(\times\) 4.6 mm empaquetada con una fase estacionaria unida a C 8 de 5 μm. La fase móvil isocrática es 37. 5:62 .5 v/v acetonitrilo y agua (que contiene 1.5 g de perclorato de tetrametilamonio y 0.1 mL de 70% v/v HClO 4). Monitoree el cromatograma usando un detector de fluorescencia ajustado a una longitud de onda de excitación de 235 nm y una longitud de onda de emisión de 310 nm.
Preguntas
1. La extracción en fase sólida es importante porque elimina constituciones en el suero que podrían interferir con el análisis. ¿Qué tipos de interferencias son posibles?
El suero sanguíneo, que es una mezcla compleja de compuestos, es aproximadamente 92% de agua, 6-8% de proteínas solubles y menos de 1% de cada una de las diversas sales, lípidos y glucosa. Una inyección directa de suero no es recomendable por tres razones. Primero, cualquier material particulado en el suero obstruirá la columna y restringirá el flujo de la fase móvil. Segundo, algunos de los compuestos en el suero pueden absorber demasiado fuertemente a la fase estacionaria, degradando el rendimiento de la columna. Finalmente, aunque una HPLC puede separar y analizar mezclas complejas, un análisis es difícil si el número de constituyentes excede la capacidad máxima de la columna.
2. Una ventaja de un análisis por HPLC es que un inyector de bucle a menudo elimina la necesidad de un estándar interno. ¿Por qué se utiliza un estándar interno en este análisis? ¿Qué suposición (s) debemos hacer al usar el estándar interno?
Un estándar interno es necesario debido a las incertidumbres introducidas durante la extracción en fase sólida. Por ejemplo, el volumen de suero transferido al cartucho de extracción en fase sólida, 0.5 mL, y el volumen de solvente utilizado para eliminar el analito y el patrón interno, 0.25 mL, son muy pequeños. La precisión y exactitud con la que podemos medir estos volúmenes no es tan buena como cuando usamos volúmenes más grandes. Por ejemplo, si extraemos el analito en un volumen de 0.24 mL en lugar de un volumen de 0.25 mL, entonces la concentración del analito aumenta ligeramente más del 4%. Además, la concentración de analitos eluidos puede variar de prueba a prueba debido a variaciones en la cantidad de solución retenida por el cartucho. El uso de un estándar interno compensa estas variaciones. Para ser útiles debemos suponer que el analito y el patrón interno se retienen completamente durante la carga inicial, que no se pierden cuando se lava el cartucho, y que se extraen completamente durante la elución final.
3. ¿Por qué el procedimiento monitorea la fluorescencia en lugar de monitorear la absorción UV?
La fluorescencia es una técnica más selectiva para detectar analitos. Muchos otros antidepresivos comúnmente recetados (y sus metabolitos) eluyen con tiempos de retención similares a los de la fluoxetina. Estos compuestos, sin embargo, o bien no fluorescen o solo son débilmente fluorescentes.
4. Si los picos de fluoxetina y protriptilina se resuelven insuficientemente, ¿cómo podría alterar la fase móvil para mejorar su separación?
Disminuir la cantidad de acetonitrilo y aumentar la cantidad de agua en el móvil aumentará los tiempos de retención, proporcionando más tiempo para efectuar una separación.
Evaluación
Con algunas excepciones, la escala de operación, precisión, precisión, sensibilidad, selectividad, tiempo de análisis y costo para un método de HPLC son similares a los métodos de GC. Los volúmenes de inyección para un método de HPLC suelen ser mayores que para un método de GC porque las columnas de HPLC tienen una mayor capacidad. Debido a que utiliza una inyección de bucle, la precisión de un método de HPLC a menudo es mejor que un método de GC. La HPLC no se limita a analitos volátiles, lo que significa que podemos analizar una gama más amplia de compuestos. Las columnas GC capilares, por otro lado, tienen placas más teóricas, y pueden separar mezclas más complejas.