
12.6: Otras Formas de Cromatografía
- Page ID
- 75656

Al inicio de la Sección 12.5, observamos que existen varios tipos diferentes de interacciones soluto/fase estacionaria en la cromatografía líquida, pero limitamos nuestra discusión a la cromatografía líquido-líquida. En esta sección volvemos nuestra atención a las técnicas de cromatografía líquida en las que el reparto ocurre por adsorción líquido-sólido, intercambio iónico y exclusión por tamaño.
Cromatografía líquido-sólido
En la cromatografía de adsorción líquido-sólido (LSC), el empaquetamiento de la columna también sirve como fase estacionaria. En el trabajo original de Tswett, la fase estacionaria estaba finamente dividida CaCo 3, pero las columnas modernas emplean partículas porosas de 3—10 μm de sílice o alúmina. Debido a que la fase estacionaria es polar, la fase móvil suele ser un disolvente no polar o moderadamente polar. Las fases móviles típicas incluyen hexano, isooctano y cloruro de metileno. El orden de vida habitual, desde tiempos de retención más cortos a más largos, es
olefinas < hidrocarburos aromáticos < éteres < ésteres, aldehídos, cetonas < alcoholes, aminas < amida < ácidos carboxílicos
También se utilizan fases estacionarias no polares, como los absorbentes a base de carbón. Para la mayoría de las muestras, la cromatografía líquido-sólido no ofrece ninguna ventaja especial sobre la cromatografía líquido-líquido. Una excepción es el análisis de isómeros, donde la LSC sobresale.
Cromatografía de intercambio iónico
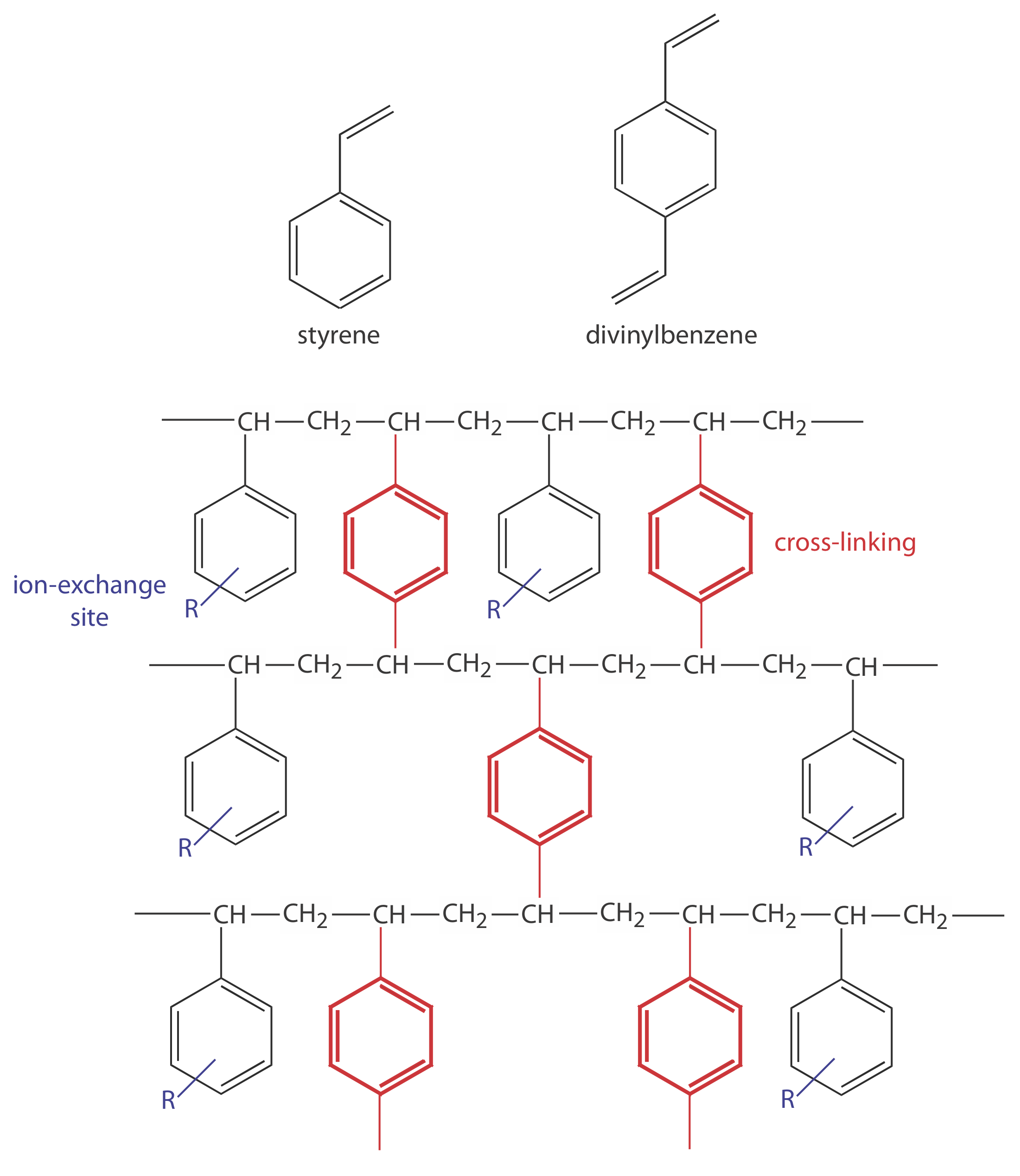
En la cromatografía de intercambio iónico (IEC) la fase estacionaria es una resina polimérica reticulada, generalmente poliestireno reticulado con divinilbenceno, con grupos funcionales iónicos unidos covalentemente (ver Figura 12.6.1 y Tabla 12.6.1 ). Los contraiones a estas cargas fijas son móviles y son desplazados por iones que compiten más favorablemente por los sitios de intercambio. Las resinas de intercambio iónico se dividen en cuatro categorías: intercambiadores catiónicos ácidos fuertes; intercambiadores catiónicos ácidos débiles; intercambiadores aniónicos de base fuerte; e intercambiadores aniónicos de base débil.
 |
 |
Figura 12.6.1 . Las estructuras de estireno, divinilbenceno y un copolímero de estireno-divinilbenceno modificado para su uso como resina de intercambio iónico se muestran a la izquierda. Los sitios de intercambio iónico, indicados por R y mostrados en azul, están en su mayoría en la posición para y no están necesariamente unidos a todas las unidades de estireno. La reticulación se muestra en rojo. La foto de la derecha muestra un ejemplo de las perlas de polímero. Estas cuentas tienen aproximadamente 0.30—0.85 mm de diámetro. Las resinas para su uso en cromatografía de intercambio iónico suelen tener un diámetro de 5—11 μm.
Los intercambiadores catiónicos de ácido fuerte incluyen un grupo funcional ácido sulfónico que lo retiene en forma aniónica y, por lo tanto, su capacidad de intercambio iónico, en soluciones fuertemente ácidas. Los grupos funcionales para un intercambiador catiónico ácido débil, por otro lado, están completamente protonados a niveles de pH inferiores a 4 y pierden su capacidad de intercambio. Los intercambiadores aniónicos de base fuerte incluyen una amina cuaternaria, que retiene una carga positiva incluso en soluciones fuertemente básicas. Los intercambiadores aniónicos de base débil permanecen protonados solo a niveles de pH moderadamente básicos. Bajo condiciones más básicas, un intercambiador aniónico de base débil pierde un protón y su capacidad de intercambio.
La reacción de intercambio iónico de un sitio de intercambio catiónico monovalente, M +, es
\[-\mathrm{SO}_{3}^{-} \mathrm{H}^{+}(s)+\mathrm{M}^{+}(a q)\rightleftharpoons-\mathrm{SO}_{3}^{-} \mathrm{M}^{+}(s)+\mathrm{H}^{+}(a q) \nonumber\]
La constante de equilibrio para esta reacción de intercambio iónico, que llamamos el coeficiente de selectividad, K, es
donde usamos llaves, {}, para indicar una concentración superficial en lugar de una concentración de solución.
No solemos pensar en la concentración de un sólido. Hay una buena razón para ello. En la mayoría de los casos, la concentración de un sólido es una constante. Si rompes un trozo de tiza en dos partes, por ejemplo, la masa y el volumen de cada pieza conservan la misma relación proporcional que en la pieza de tiza original. Sin embargo, cuando consideramos una unión iónica a un sitio reactivo en la superficie del sólido, la fracción de sitios que están unidos, y por lo tanto la concentración de sitios unidos, puede tomar cualquier valor entre 0 y algún valor máximo que sea proporcional a la densidad de sitios reactivos.
Reordenando la ecuación\ ref {12.1} nos muestra que la relación de distribución, D, para la reacción de intercambio
\[D=\frac{\text { amount of } \mathrm{M}^{+} \text { in the stationary phase }}{\text { amount of } \mathrm{M}^{+} \text { in the mobile phase }} \nonumber\]
es una función de la concentración de H + y, por tanto, del pH de la fase móvil.
La selectividad de una resina de intercambio iónico depende en cierta medida de si incluye sitios de intercambio fuertes o débiles y del grado de reticulación. Esto último es particularmente importante ya que controla la permeabilidad de la resina y, por lo tanto, la accesibilidad de los sitios de intercambio. Un orden aproximado de selectividad para una resina de intercambio catiónico de ácido fuerte típica, en orden decreciente de D, es
Al 3 + > Ba 2+ > Pb 2 + > Ca 2 + > Ni 2 + > Cd 2 + > Cu 2 + > Co 2 + > Zn 2 + > Mg 2 + > Ag + > K + >\(\text{NH}_4^+\) > Na + > H + > Li +
Obsérvese que los cationes altamente cargados se unen más fuertemente que los cationes de menor carga, y que para los cationes de carga similar, aquellos con un radio hidratado menor (ver Cuadro 6.9.1 en el Capítulo 6), o que son más polarizables, se unen más fuertemente. Para un intercambiador aniónico de base fuerte, el orden general de elución es
\(\text{SO}_4^{2-}\)> I — >\(\text{HSO}_4^-\) >\(\text{NO}_3^-\) > Br —\(\text{NO}_2^-\) > > Cl — >\(\text{HCO}_3^-\) > CH3COO — > OH — > F —
Los aniones de mayor carga y de menor radio hidratado se unen con mayor fuerza que los aniones con menor carga y mayor radio hidratado.
La fase móvil en IEC suele ser un tampón acuoso, cuyo pH y composición iónica determinan el tiempo de retención de un soluto. Las eluciones en gradiente son posibles en las que la fuerza iónica o el pH de la fase móvil se cambia con el tiempo. Por ejemplo, una separación IEC de cationes podría usar una solución diluida de HCl como fase móvil. El aumento de la concentración de HCl acelera la tasa de elución para cationes retenidos más fuertemente porque la mayor concentración de H + le permite competir con mayor éxito por los sitios de intercambio iónico.
A partir de la Ecuación\ ref {12.2}, la relación de distribución de un catión, D, se vuelve más pequeña cuando aumenta la concentración de H + en la fase móvil.
Una resina de intercambio iónico se incorpora a una columna de HPLC ya sea como perlas de polímero poroso de 5—11 μm o recubriendo la resina sobre partículas porosas de sílice. Las columnas suelen tener una longitud de 250 mm con diámetros internos que van de 2 a 5 mm.
La medición de la conductividad de la fase móvil a medida que eluye de la columna sirve como detector universal para analitos catiónicos y aniónicos. Debido a que la fase móvil contiene una alta concentración de iones —una fase móvil de HCl diluido, por ejemplo, contiene concentraciones significativas de H + y Cl —, necesitamos un método para detectar los analitos en presencia de una conductividad de fondo significativa.
Para minimizar la contribución de la fase móvil a la conductividad, se coloca una columna supresora de iones entre la columna analítica y el detector. Esta columna elimina selectivamente iones de fase móvil sin eliminar iones soluto. Por ejemplo, en cromatografía de intercambio catiónico usando una solución diluida de HCl como fase móvil, la columna supresora contiene una resina de intercambio aniónico de base fuerte. La reacción de intercambio
\[\mathrm{H}^{+}(a q)+\mathrm{Cl}^{-}(a q)+\mathrm{Resin}^{+} \mathrm{OH}^{-}(s)\rightleftharpoons\operatorname{Resin}^{+} \mathrm{Cl}^{-}(s)+\mathrm{H}_{2} \mathrm{O}(l ) \nonumber\]
reemplaza los iones de fase móvil H + y Cl — por H 2 O. Un proceso similar se utiliza en la cromatografía de intercambio aniónico donde la columna supresora contiene una resina de intercambio catiónico. Si la fase móvil es una solución de Na 2 CO 3, la reacción de intercambio
\[2 \mathrm{Na}^{+}(a q)+\mathrm{CO}_{3}^{2-}(a q)+2 \operatorname{Resin}^{-} \mathrm{H}^{+}(s)\rightleftharpoons2 \operatorname{Resin}^{-} \mathrm{Na}^{+}(s)+\mathrm{H}_{2} \mathrm{CO}_{3}(a q) \nonumber\]
reemplaza un electrolito fuerte, Na 2 CO 3, por un electrolito débil, H 2 CO 3.
La supresión de iones es necesaria cuando la fase móvil contiene una alta concentración de iones. La cromatografía iónica de una sola columna, en la que no se necesita una columna supresora de iones, es posible si la concentración de iones en la fase móvil es pequeña. Típicamente, la fase estacionaria es una resina con una baja capacidad de intercambio iónico y la fase móvil es una solución muy diluida de ácido metansulfónico para analitos catiónicos, o benzoato de potasio o hidrógeno ftalato de potasio para analitos aniónicos. Debido a que la conductividad de fondo es suficientemente pequeña, es posible monitorear un cambio en la conductividad a medida que los analitos eluyen de la columna.
Se puede usar un detector de absorbancia UV/Vis si los analitos absorben radiación ultravioleta o visible. Alternativamente, podemos detectar indirectamente analitos que no absorben en los UV/Vis si la fase móvil contiene una especie absorbente de UV/Vis. En este caso, cuando una banda de soluto pasa a través del detector, se mide una disminución en la absorbancia en el detector.
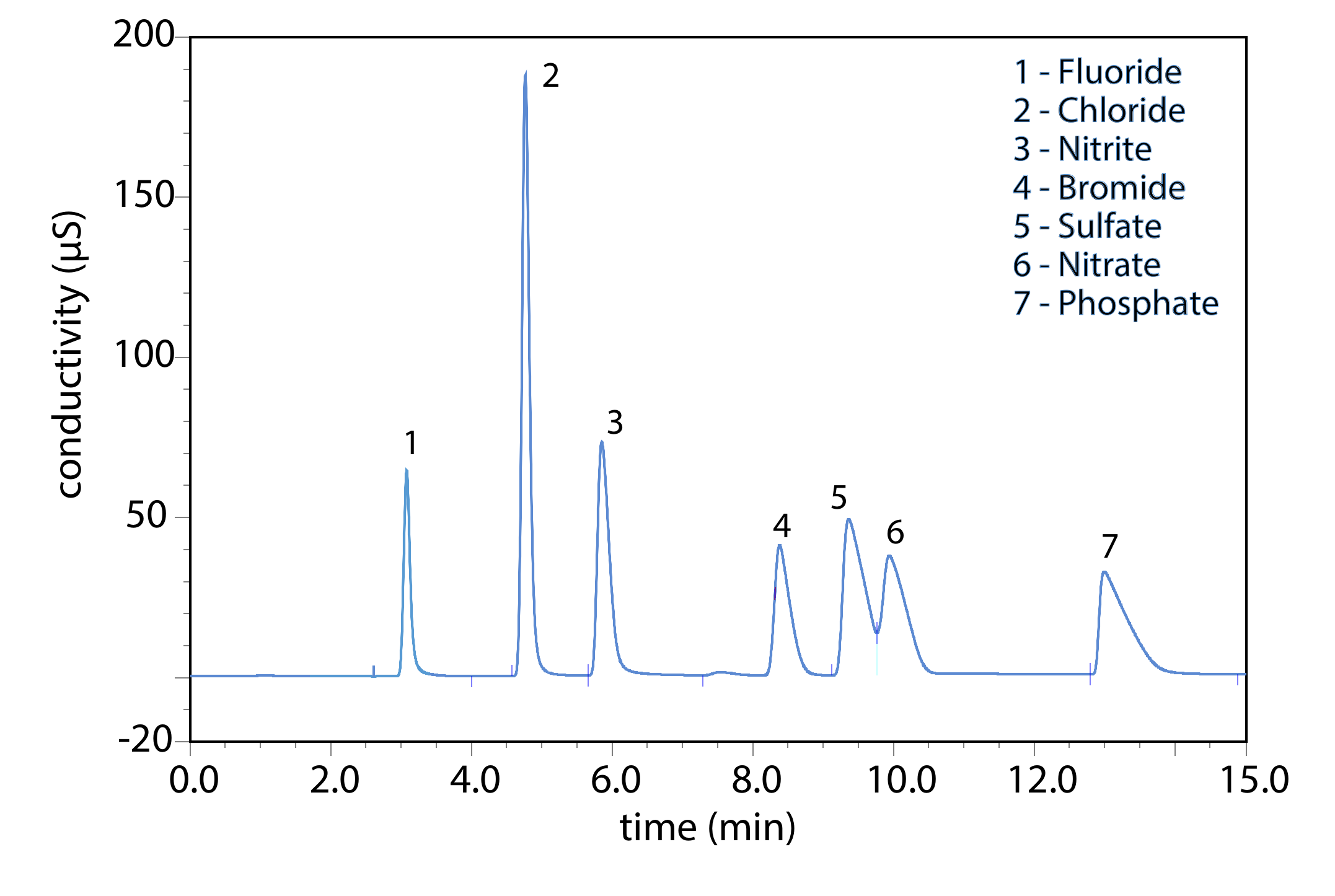
La cromatografía de intercambio iónico es una técnica importante para el análisis de aniones y cationes en agua. Por ejemplo, un análisis cromatográfico de intercambio iónico para los aniones F —, Cl —, Br —\(\text{NO}_2^-\),\(\text{NO}_3^-\),\(\text{PO}_4^{3-}\), y\(\text{SO}_4^{2-}\) toma aproximadamente 15 minutos (Figura 12.6.2 ). Un análisis completo del mismo conjunto de aniones mediante una combinación de potenciometría y espectrofotometría requiere de 1 a 2 días. La cromatografía de intercambio iónico también se utiliza para el análisis de proteínas, aminoácidos, azúcares, nucleótidos, productos farmacéuticos, productos de consumo y muestras clínicas.
Cromatografía de exclusión por tamaño
Hemos considerado dos clases de fases estacionarias de tamaño micrométrico en esta sección: partículas de sílice y perlas de resina de polímero reticulado. Ambos materiales son porosos, con tamaños de poro que van desde aproximadamente 5—400 nm para partículas de sílice, y de 5 nm a 100 μm para resinas de poliestireno reticulado con divinilbenceno. En la cromatografía de exclusión por tamaño, que también se conoce por los términos cromatografía de exclusión molecular o de permeación en gel, la separación de solutos depende de su capacidad para entrar en los poros de la fase estacionaria. Los solutos más pequeños pasan proporcionalmente más tiempo dentro de los poros y tardan más en eluirse de la columna.
La selectividad de tamaño de una fase estacionaria se extiende sobre un rango finito. Todos los solutos significativamente más pequeños que los poros se mueven a través de todo el volumen de la columna y eluyen simultáneamente, con un volumen de retención, V r, de
\[V_{r}=V_{i}+V_{o} \label{12.3}\]
donde V i es el volumen de la fase móvil que ocupa el espacio de poro de la fase estacionaria y V o es el volumen de la fase móvil en el resto de la columna. El soluto más grande para el que se mantiene la Ecuación\ ref {12.3} es el límite de inclusión de la columna, o límite de permeación. Esos solutos demasiado grandes para entrar en los poros eluyen simultáneamente con un volumen de retención de
\[V_{r} = V_{o} \label{12.4}\]
La ecuación\ ref {12.4} define el límite de exclusión de la columna.
Para un soluto cuyo tamaño está entre el límite de inclusión y el límite de exclusión, la cantidad de tiempo que pasa en los poros de la fase estacionaria es proporcional a su tamaño. El volumen de retención para estos solutos es
\[V_{r}=DV_{i}+V_{o} \label{12.5}\]
donde D es la relación de distribución del soluto, que varía de 0 en el límite de exclusión a 1 en el límite de inclusión. La ecuación\ ref {12.5} asume que la exclusión por tamaño es la única interacción entre el soluto y la fase estacionaria que afecta a la separación. Por esta razón, las fases estacionarias que utilizan partículas de sílice se desactivan como se describió anteriormente, y las resinas poliméricas se sintetizan sin sitios de intercambio.
La cromatografía de exclusión por tamaño proporciona un medio rápido para separar moléculas más grandes, incluyendo polímeros y biomoléculas. Una fase estacionaria para proteínas que consiste en partículas con poros de 30 nm tiene un límite de inclusión de 7500 g/mol y un límite de exclusión de\(1.2 \times 10^6\) g/mol. Las mezclas de proteínas que abarcan un rango más amplio de pesos moleculares se separan uniendo en serie varias columnas con diferentes límites de inclusión y exclusión.
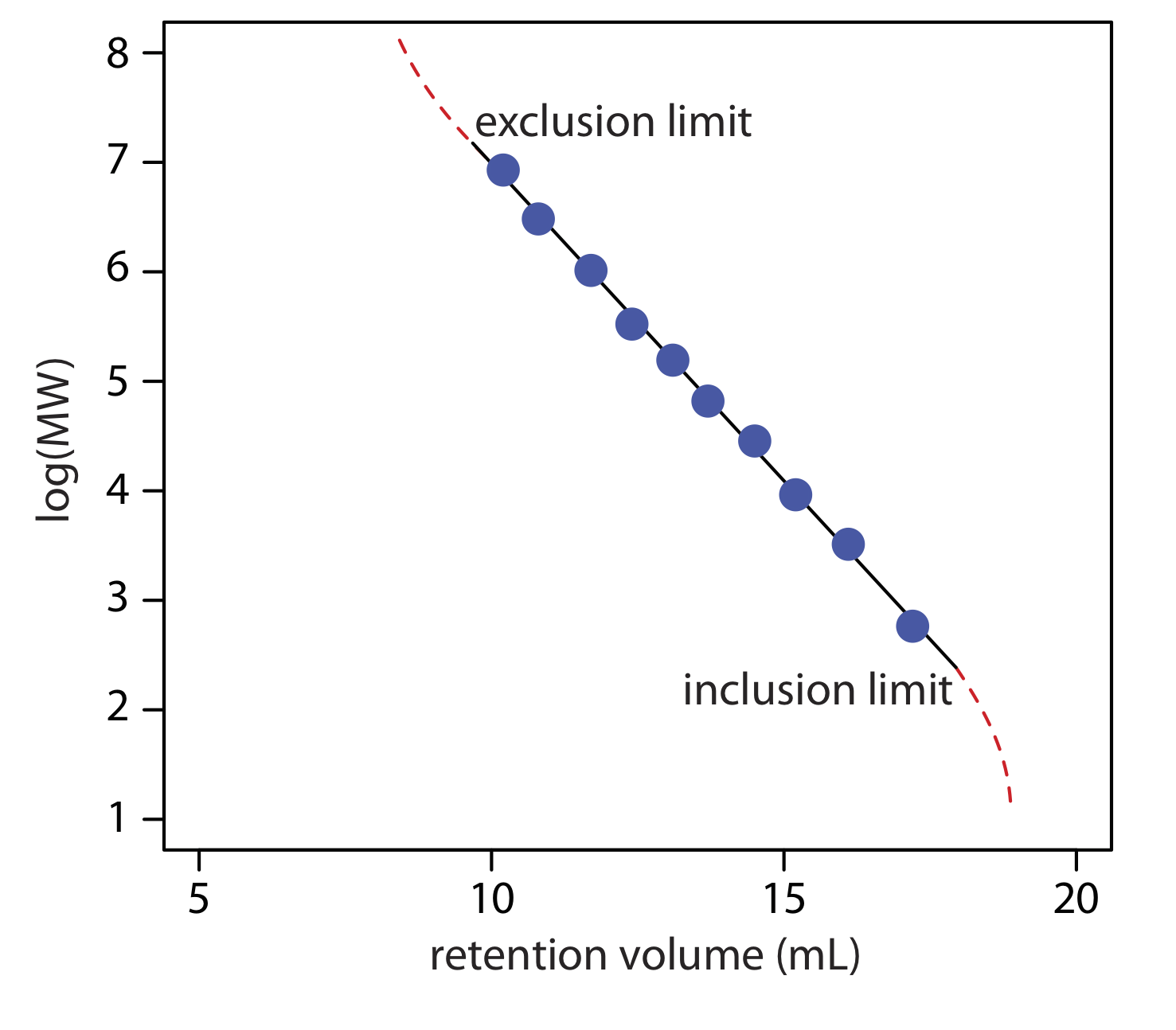
Otra aplicación importante de la cromatografía de exclusión por tamaño es la estimación del peso molecular (PM) de un soluto. Las curvas de calibración se preparan usando una serie de patrones de peso molecular conocido y midiendo el volumen de retención de cada patrón. Como se muestra en la Figura 12.6.3 , una gráfica de log (MW) versus V r es aproximadamente lineal entre el límite de exclusión y el límite de inclusión. Debido a que el volumen de retención de un soluto está influenciado tanto por su tamaño como por su forma, una estimación razonablemente precisa del peso molecular solo es posible si los estándares se eligen cuidadosamente para minimizar el efecto de la forma.
La cromatografía de exclusión por tamaño se lleva a cabo utilizando instrumentación HPLC convencional, reemplazando la columna HPLC por una columna de exclusión por tamaño apropiada. Un detector UV/Vis es el medio más común para obtener el cromatograma.
Cromatografía de fluidos supercríticos
Si bien existen muchas aplicaciones analíticas de cromatografía de gases y cromatografía líquida, no pueden separar y analizar todo tipo de muestras. La columna capilar GC separa mezclas complejas con excelente resolución y tiempos de análisis cortos. Su aplicación se limita, sin embargo, a analitos volátiles o a analitos que se vuelven volátiles mediante una reacción de derivatización adecuada. La cromatografía líquida separa una gama más amplia de solutos que la GC, pero los detectores más comunes (UV, fluorescencia y electroquímicos) tienen límites de detección más pobres y rangos lineales más pequeños que los detectores de GC, y no son tan universales en su selectividad.
Para algunas muestras, la cromatografía de fluidos supercríticos (SFC) proporciona una alternativa útil a la cromatografía de gases y la cromatografía líquida. La fase móvil en la cromatografía de fluidos supercríticos es un gas mantenido a una temperatura y presión que excede su punto crítico (Figura 12.6.4 ). En estas condiciones la fase móvil no es ni un gas ni un líquido. En cambio, la fase móvil es un fluido supercrítico.
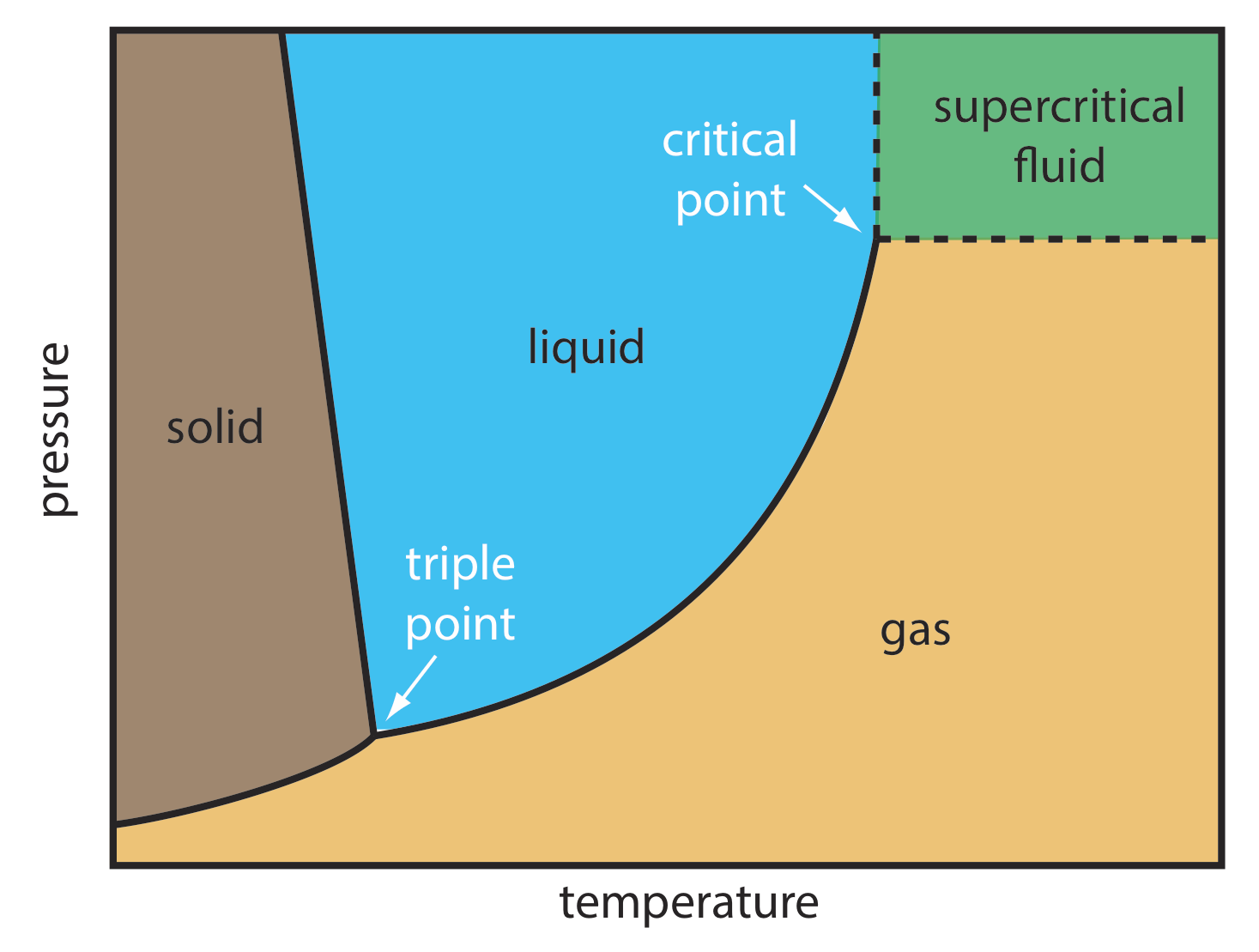
Algunas propiedades de un fluido supercrítico, como se muestra en la Tabla 12.6.2 , son similares a un gas; otras propiedades, sin embargo, son similares a un líquido. La viscosidad de un fluido supercrítico, por ejemplo, es similar a la de un gas, lo que significa que podemos mover un fluido supercrítico a través de una columna capilar o una columna empaquetada sin las altas presiones necesarias en HPLC. El tiempo de análisis y la resolución, aunque no tan buenos como en GC, suelen ser mejores que en HPLC convencional. La densidad de un fluido supercrítico, por otro lado, es mucho más cercana a la de un líquido, lo que explica por qué los fluidos supercríticos son buenos solventes. En cuanto a su poder de separación, una fase móvil en SFC se comporta más como la fase móvil líquida en HPLC que la fase móvil gaseosa en GC.
La fase móvil más común para la cromatografía de fluidos supercríticos es el CO 2. Su baja temperatura crítica de 31.1 o C y su baja presión crítica de 72.9 atm son relativamente fáciles de lograr y mantener. Aunque el CO 2 supercrítico es un buen disolvente para los orgánicos no polares, es menos útil para los solutos polares. La adición de un modificador orgánico, tal como metanol, mejora la fuerza de elución de la fase móvil. Otras fases móviles comunes y sus temperaturas y presiones críticas se enumeran en la Tabla 12.6.3 .
La instrumentación para cromatografía de fluidos supercríticos es esencialmente la misma que para una HPLC estándar. Las únicas adiciones importantes son un horno calentado para la columna y un limitador de presión aguas abajo de la columna para mantener la presión crítica. Las eluciones de gradiente se logran cambiando la presión aplicada a lo largo del tiempo. El cambio resultante en la densidad de la fase móvil afecta su fuerza de solvente. La detección se realiza usando detectores de GC estándar o detectores de HPLC. La cromatografía de fluidos supercríticos tiene muchas aplicaciones en el análisis de polímeros, combustibles fósiles, ceras, medicamentos y productos alimenticios.