
12.7: Electroforesis
- Page ID
- 75639

La electroforesis es una clase de técnicas de separación en la que separamos analitos por su capacidad de moverse a través de un medio conductor, generalmente un tampón acuoso, en respuesta a un campo eléctrico aplicado. En ausencia de otros efectos, los cationes migran hacia el cátodo cargado negativamente del campo eléctrico. Los cationes con mayores proporciones de carga a tamaño, lo que favorece los iones de mayor carga y de menor tamaño, migran a una velocidad más rápida que los iones cat más grandes con cargas más pequeñas. Los aniones migran hacia el ánodo cargado positivamente y las especies neutras no experimentan el campo eléctrico y permanecen estacionarias.
Como veremos en breve, en condiciones normales incluso especies neutras y aniones migran hacia el cátodo.
Existen varias formas de electroforesis. En la electroforesis en gel en placa, el tampón conductor se retiene dentro de un gel poroso de agarosa o poliacrilamida. Las losas se forman vertiendo el gel entre dos placas de vidrio separadas por espaciadores. Los espesores típicos son 0.25—1 mm. La electroforesis en gel es una técnica importante en bioquímica donde se utiliza frecuentemente para separar fragmentos de ADN y proteínas. Si bien es una herramienta poderosa para el análisis cualitativo de mezclas complejas, es menos útil para el trabajo cuantitativo.
En la electroforesis capilar, el tampón conductor se retiene dentro de un tubo capilar con un diámetro interno que típicamente es de 25—75 μm. La muestra se inyecta en un extremo del tubo capilar, y a medida que migra a través del capilar los componentes de la muestra se separan y eluyen de la columna en diferentes momentos. El electroferograma resultante se parece a un cromatograma GC o HPLC, y proporciona información tanto cualitativa como cuantitativa. Solo los métodos electroforéticos capilares reciben mayor consideración en esta sección.
Teoría de Electroforesis
En electroforesis capilar inyectamos la muestra en una solución tamponada retenida dentro de un tubo capilar. Cuando se aplica un campo eléctrico a través del tubo capilar, los componentes de la muestra migran como resultado de dos tipos de acciones: movilidad electroforética y movilidad electroosmótica. La movilidad electroforética es la respuesta del soluto al campo eléctrico aplicado en el que los cationes se mueven hacia el cátodo cargado negativamente, los aniones se mueven hacia el ánodo cargado positivamente y las especies neutras permanecen estacionarias. La otra contribución a la migración de un soluto es el flujo electroosmótico, que ocurre cuando el tampón se mueve a través del capilar en respuesta al campo eléctrico aplicado. En condiciones normales el tampón se mueve hacia el cátodo, barriendo la mayoría de los solutos, incluyendo los aniones y especies neutras, hacia el cátodo cargado negativamente.
Movilidad Electroforética
La velocidad con la que se mueve un soluto en respuesta al campo eléctrico aplicado se denomina velocidad electroforética,\(\nu_{ep}\); se define como
\[\nu_{ep}=\mu_{ep} E \label{12.1}\]
donde\(\mu_{ep}\) está la movilidad electroforética del soluto, y E es la magnitud del campo eléctrico aplicado. La movilidad electroforética de un soluto se define como
\[\mu_{ep}=\frac{q}{6 \pi \eta r} \label{12.2}\]
donde q es la carga del soluto,\(\eta\) es la viscosidad del tampón y r es el radio del soluto. Usando la Ecuación\ ref {12.1} y la Ecuación\ ref {12.2} podemos sacar varias conclusiones importantes sobre la velocidad electroforética de un soluto. La movilidad electroforética y, por lo tanto, la velocidad electroforética, aumenta para solutos con mayor carga y para solutos de menor tamaño. Debido a que q es positivo para un catión y negativo para un anión, estas especies migran en direcciones opuestas. Una especie neutra, para la cual q es cero, tiene una velocidad electroforética de cero.
Movilidad Electroosmótica
Cuando se aplica un campo eléctrico a un capilar lleno de un tampón acuoso, esperamos que los iones del tampón migren en respuesta a su movilidad electroforética. Debido a que el disolvente, H 2 O, es neutro, podríamos esperar razonablemente que permanezca estacionario. Lo que observamos en condiciones normales, sin embargo, es que el búfer se mueve hacia el cátodo. A este fenómeno se le llama flujo electroosmótico.
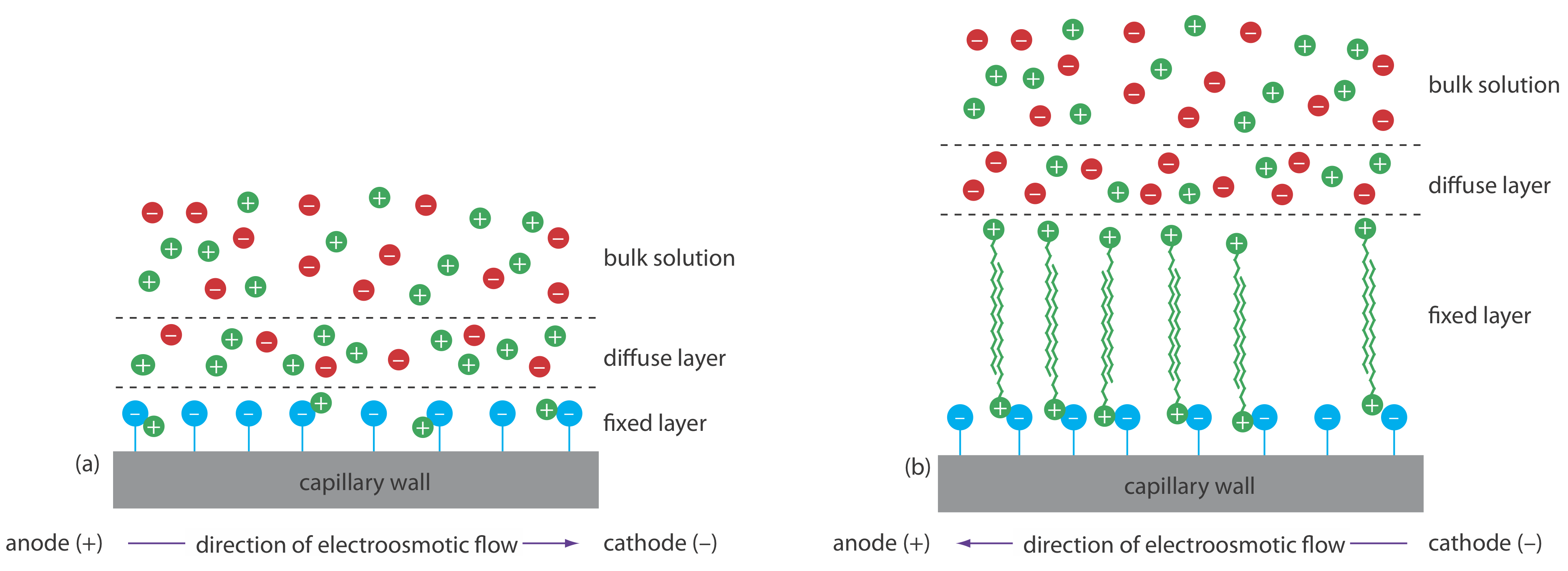
El flujo electroosmótico ocurre porque las paredes del tubo capilar transportan una carga. La superficie de un capilar de sílice contiene un gran número de grupos silanol (—SiOH). A un nivel de pH superior a aproximadamente 2 o 3, los grupos silanol se ionizan para formar iones de silanato cargados negativamente (—SiO —). Los cationes del tampón son atraídos por los iones de silanato. Como se muestra en la Figura 12.7.1 , algunos de estos cationes se unen fuertemente a los iones silanato, formando una capa fija. Debido a que los cationes en la capa fija solo neutralizan parcialmente la carga negativa en las paredes capilares, la solución adyacente a la capa fija, que se llama capa difusa, contiene más cationes que aniones. Juntas estas dos capas se conocen como la doble capa. Los cationes en la capa difusa migran hacia el cátodo. Debido a que estos cationes están solvatados, la solución también es arrastrada, produciendo el flujo electroosmótico.
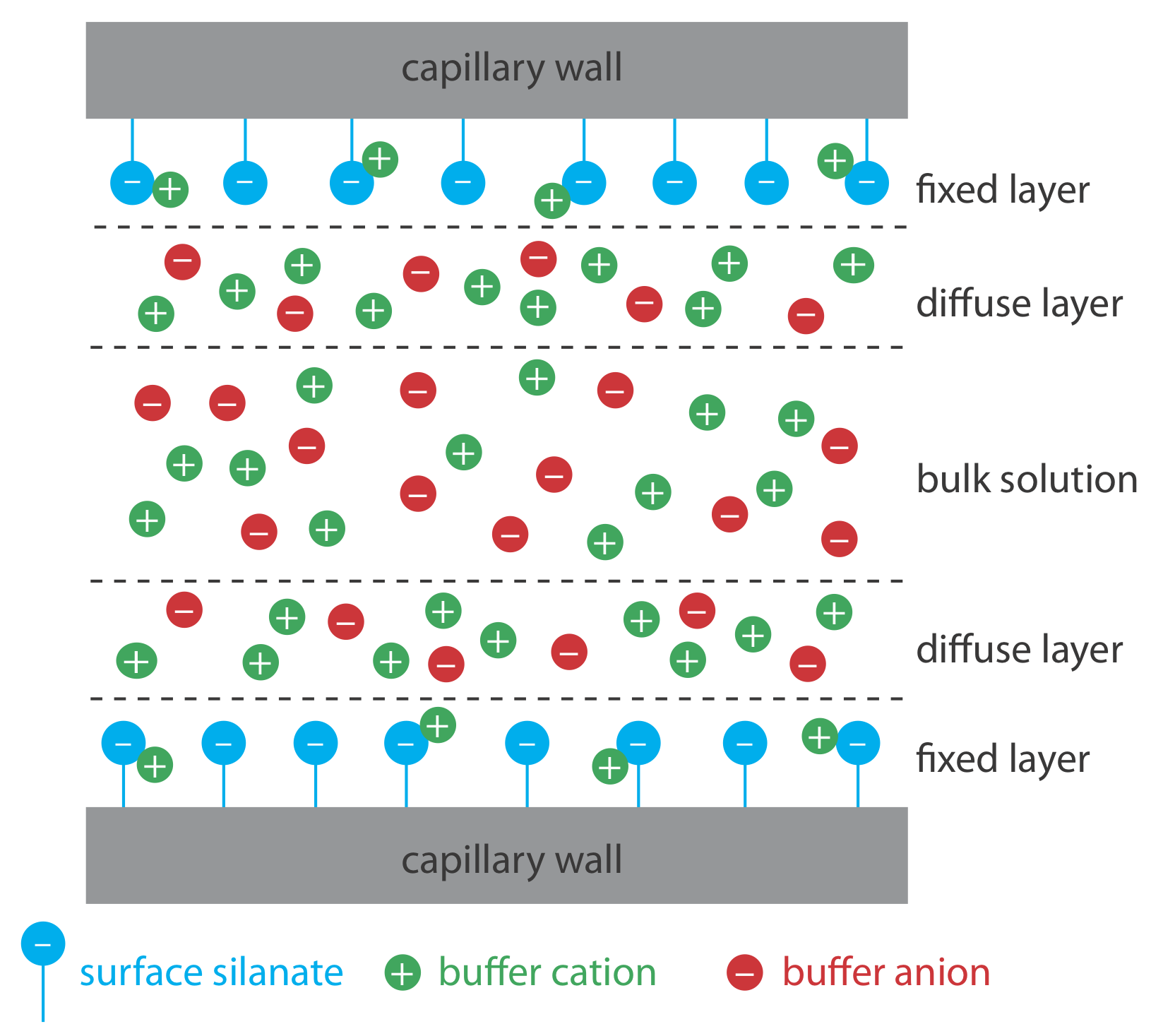
Los aniones en la capa difusa, que también están solvatados, intentan moverse hacia el ánodo. Debido a que hay más cationes que aniones, sin embargo, los cationes ganan y el flujo electroosmótico se mueve en la dirección del cátodo.
La velocidad a la que el búfer se mueve a través del capilar, lo que llamamos su velocidad de flujo electroosmótico\(\nu_{eof}\),, es una función del campo eléctrico aplicado, E, y la movilidad electroosmótica del búfer,\(\mu_{eof}\).
\[\nu_{eof}=\mu_{e o f} E \label{12.3}\]
La movilidad electroosmótica se define como
\[\mu_{eof}=\frac{\varepsilon \zeta}{4 \pi \eta} \label{12.4}\]
donde\(\epsilon\) es la constante dieléctrica del tampón,\(\zeta\) es el potencial zeta, y\(\eta\) es la viscosidad del tampón.
El potencial zeta —el potencial de la capa difusa a una distancia finita de la pared capilar— juega un papel importante en la determinación de la velocidad del flujo electroosmótico. Dos factores determinan el valor del potencial zeta. Primero, el potencial zeta es directamente proporcional a la carga en las paredes capilares, con una mayor densidad de iones silanato que corresponde a un mayor potencial zeta. Por debajo de un pH de 2 hay pocos iones de silanato y el potencial zeta y la velocidad de flujo electroosmótico se acercan a cero. A medida que aumenta el pH, tanto el potencial zeta como la velocidad de flujo electroosmótico aumentan. Segundo, el potencial zeta es directamente proporcional al grosor de la doble capa. El aumento de la fuerza iónica del tampón proporciona una mayor concentración de cationes, lo que disminuye el grosor de la doble capa y disminuye el flujo electroosmótico.
La definición de potencial zeta dada aquí es cierto que es un poco borrosa. Para una explicación más detallada ver Delgado, A. V.; González-Caballero, F.; Hunter, R. J.; Koopal, L. K.; Lyklema, J. “Medición e Interpretación de Fenómenos Electrocinéticos”, Puro. Appl. Chem. 2005, 77, 1753—1805. Si bien se trata de un informe muy técnico, las Secciones 1.3—1.5 proporcionan una buena introducción a la dificultad de definir el potencial zeta y de medir su valor.
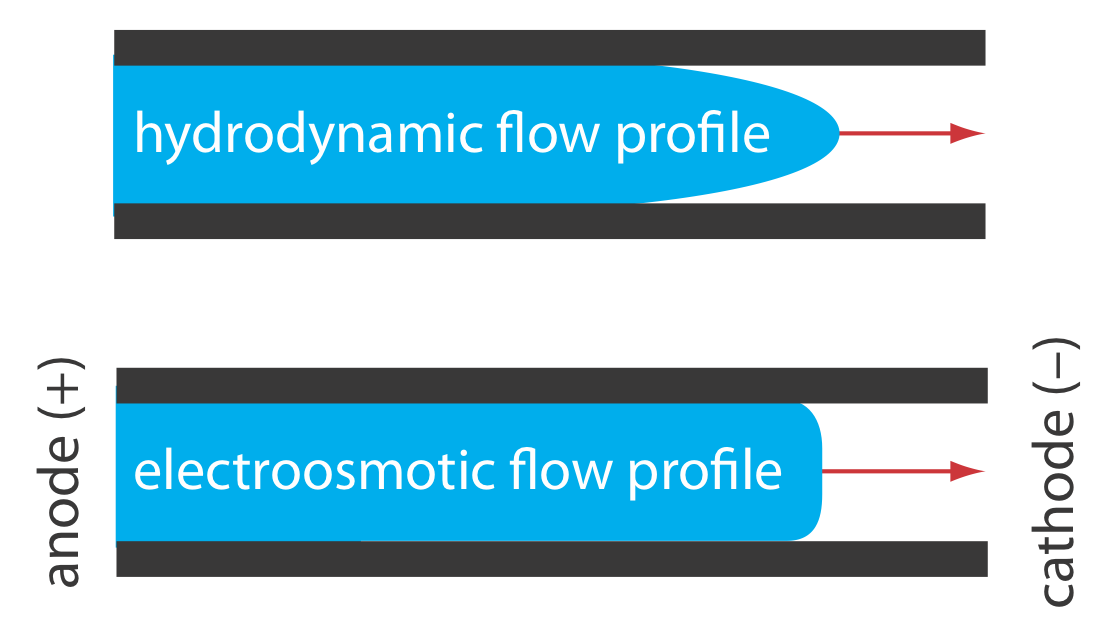
El perfil de flujo electroosmótico es muy diferente al de un fluido que se mueve bajo presión forzada. La Figura 12.7.2 compara el perfil de flujo electroosmótico con el perfil de flujo hidrodinámico en cromatografía de gases y cromatografía líquida. El perfil uniforme y plano para electroósmosis ayuda a minimizar el ensanchamiento de banda en la electroforesis capilar, mejorando la eficiencia de separación.
Movilidad Total
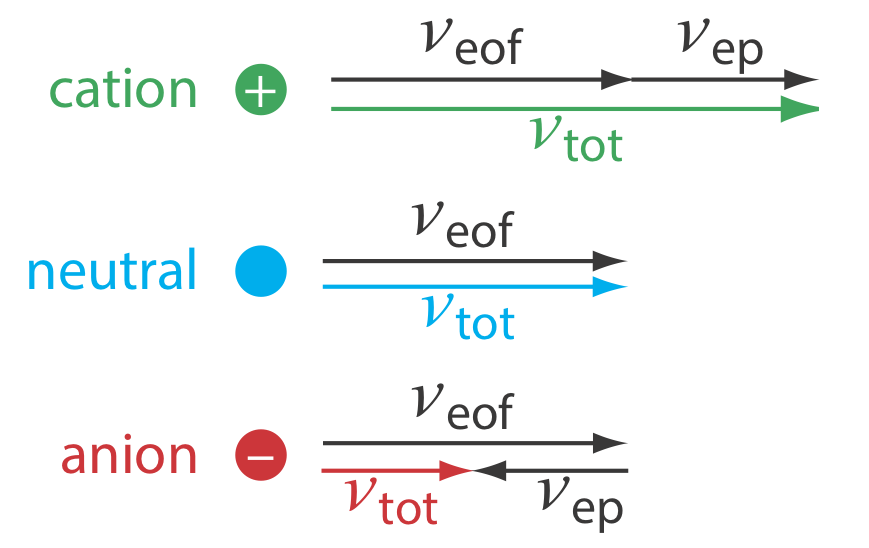
La velocidad total de un soluto\(\nu_{tot}\), a medida que se mueve a través del capilar es la suma de su velocidad electroforética y la velocidad de flujo electroosmótico.
\[\nu_{t o t}=\nu_{ep}+\nu_{eof} \nonumber\]
Como se muestra en la Figura 12.7.3 , en condiciones normales las siguientes relaciones generales se mantienen verdaderas.
\[(\nu_{tot})_{cations} > \nu_{eof} \nonumber\]
\[(\nu_{tot})_{neutrals} = \nu_{eof} \nonumber\]
\[(\nu_{tot})_{anions} < \nu_{eof} \nonumber\]
Los cationes eluyen primero en un orden que corresponde a sus movilidades electroforéticas, con cationes pequeños y altamente cargados eluyendo antes de cationes más grandes de menor carga. Las especies neutras eluyen como una sola banda con una tasa de elución igual a la velocidad de flujo electroosmótico. Finalmente, los aniones son los últimos componentes en eluir, con los aniones más pequeños y altamente cargados que tienen el tiempo de elución más largo.
Tiempo de migración
Otra forma de expresar la velocidad de un soluto es dividir la distancia que recorre por el tiempo transcurrido
\[\nu_{tot}=\frac{l}{t_{m}} \label{12.5}\]
donde l es la distancia entre el punto de inyección y el detector, y t m es el tiempo de migración del soluto. Para comprender las variables experimentales que afectan el tiempo de migración, comenzamos por señalar que
\[\nu_{tot} = \mu_{tot}E = (\mu_{ep} + \mu_{eof})E \label{12.6}\]
Combinando la Ecuación\ ref {12.5} y la Ecuación\ ref {12.6} y resolviendo para t m nos deja con
\[t_{\mathrm{m}}=\frac{l}{\left(\mu_{ep}+\mu_{eof}\right) E} \label{12.7}\]
La magnitud del campo eléctrico es
\[E=\frac{V}{L} \label{12.8}\]
donde V es el potencial aplicado y L es la longitud del tubo capilar. Finalmente, sustituyendo la Ecuación\ ref {12.8} por la Ecuación\ ref {12.7} nos deja con la siguiente ecuación para el tiempo de migración de un soluto.
\[t_{\mathrm{m}}=\frac{lL}{\left(\mu_{ep}+\mu_{eof}\right) V} \label{12.9}\]
Para disminuir el tiempo de migración de un soluto, lo que acorta el tiempo de análisis, podemos aplicar un voltaje más alto o usar un tubo capilar más corto. También podemos acortar el tiempo de migración aumentando el flujo electroosmótico, aunque esto disminuye la resolución.
Eficiencia
Como aprendimos en el Capítulo 12.2, la eficiencia de una separación viene dada por el número de placas teóricas, N. En electroforesis capilar el número de placas teóricas es
\[N=\frac{l^{2}}{2 D t_{m}}=\frac{\left(\mu_{e p}+\mu_{eof}\right) E l}{2 D L} \label{12.10}\]
donde D es el coeficiente de difusión del soluto. A partir de la Ecuación\ ref {12.10}, la eficiencia de una separación electroforética capilar aumenta con mayores voltajes. Aumentar la velocidad de flujo electroosmótico mejora la eficiencia, pero a expensas de la resolución. Dos observaciones adicionales merecen comentario. Primero, los solutos con mayores movilidades electroforéticas —en la misma dirección que el flujo electroosmótico— tienen mayores eficiencias; así, los cationes más pequeños y con mayor carga no son solo los primeros solutos en eluir, sino que lo hacen con mayor eficiencia. Segundo, la eficiencia en la electroforesis capilar es independiente de la longitud del capilar. Los recuentos teóricos de placas de aproximadamente 100 000—200 000 no son inusuales.
Es posible diseñar un experimento electroforético para que los aniones eluyan antes de los cationes, más sobre esto más tarde, en el que los aniones más pequeños y altamente cargados eluyen con mayores eficiencias.
Selectividad
En cromatografía definimos la selectividad entre dos solutos como la relación de sus factores de retención. En electroforesis capilar, la expresión análoga para la selectividad es
\[\alpha=\frac{\mu_{ep, 1}}{\mu_{ep, 2}} \nonumber\]
donde\(\mu_{ep,1}\) y\(\mu_{ep,2}\) son las movilidades electroforéticas para los dos solutos, elegidas de tal manera que\(\alpha \ge 1\). A menudo podemos mejorar la selectividad ajustando el pH de la solución tampón. Por ejemplo,\(\text{NH}_4^+\) es un ácido débil con una p K a de 9.75. A un pH de 9.75 las concentraciones de\(\text{NH}_4^+\) y NH 3 son iguales. Disminuir el pH por debajo de 9.75 aumenta su movilidad electroforética debido a que una mayor fracción del soluto está presente como catión\(\text{NH}_4^+\). Por otro lado, al elevar el pH por encima de 9.75 se incrementa la proporción de NH 3 neutro, disminuyendo su movilidad electroforética.
Resolución
La resolución entre dos solutos es
donde\(\mu_{avg}\) es la movilidad electroforética promedio para los dos solutos. Aumentar el voltaje aplicado y disminuir la velocidad de flujo electroosmótico mejora la resolución. Este último efecto es particularmente importante. Aunque aumentar el flujo electroosmótico mejora el tiempo de análisis y la eficiencia, disminuye la resolución.
Instrumentación
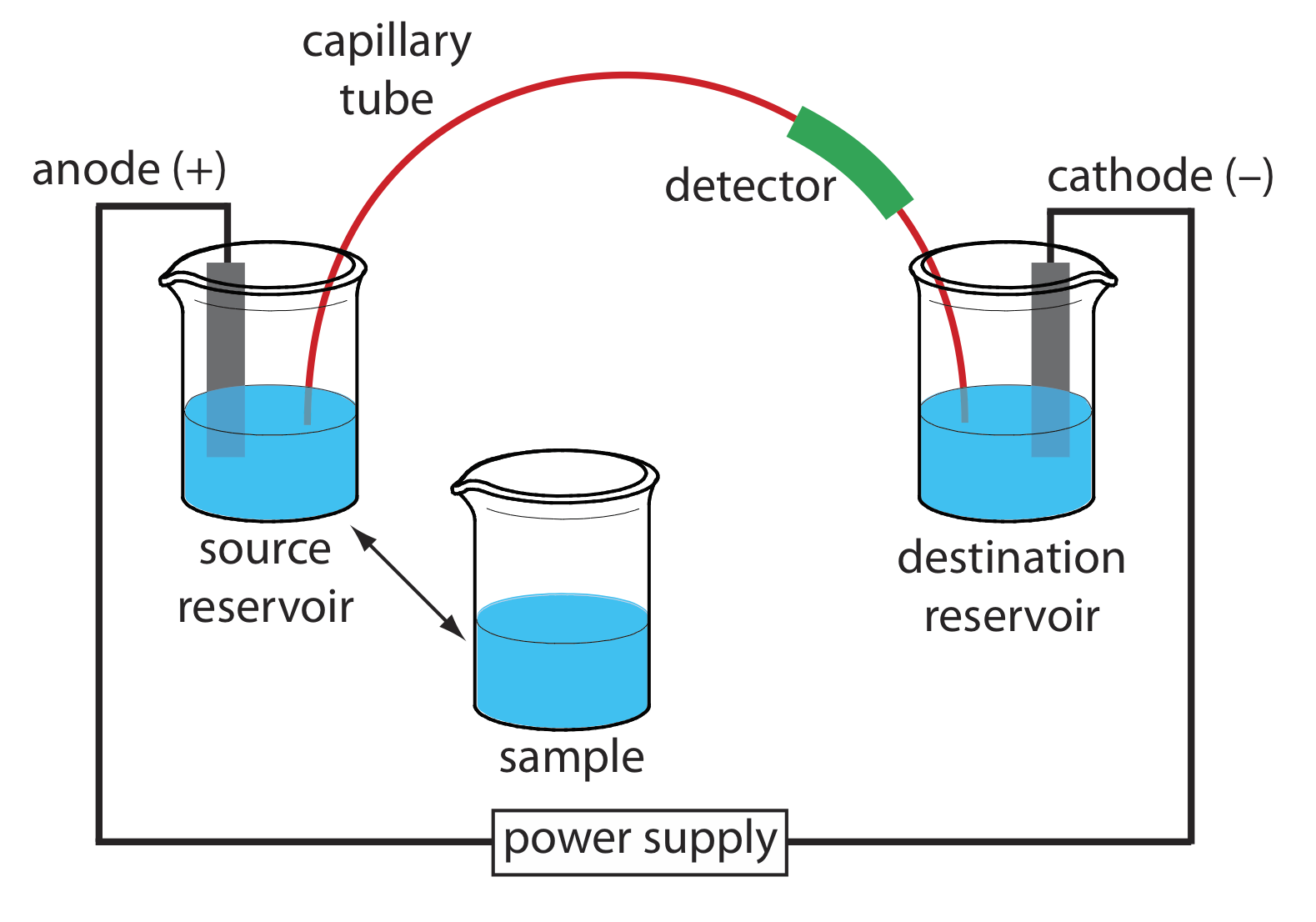
La instrumentación básica para electroforesis capilar se muestra en la Figura 12.7.4 e incluye una fuente de alimentación para aplicar el campo eléctrico, los compartimientos de ánodo y cátodo que contienen depósitos de la solución tampón, un vial de muestra que contiene la muestra, el tubo capilar y un detector . Cada parte del instrumento recibe mayor consideración en esta sección.
Tubos Capilares
La Figura 12.7.5 muestra una sección transversal de un tubo capilar típico. La mayoría de los tubos capilares están hechos de sílice fundida recubierta con una capa de 15—35 μm de poliimida para darle resistencia mecánica. El diámetro interno es típicamente de 25—75 μm, que es más pequeño que el diámetro interno de una columna capilar GC, con un diámetro exterior de 200—375 μm.
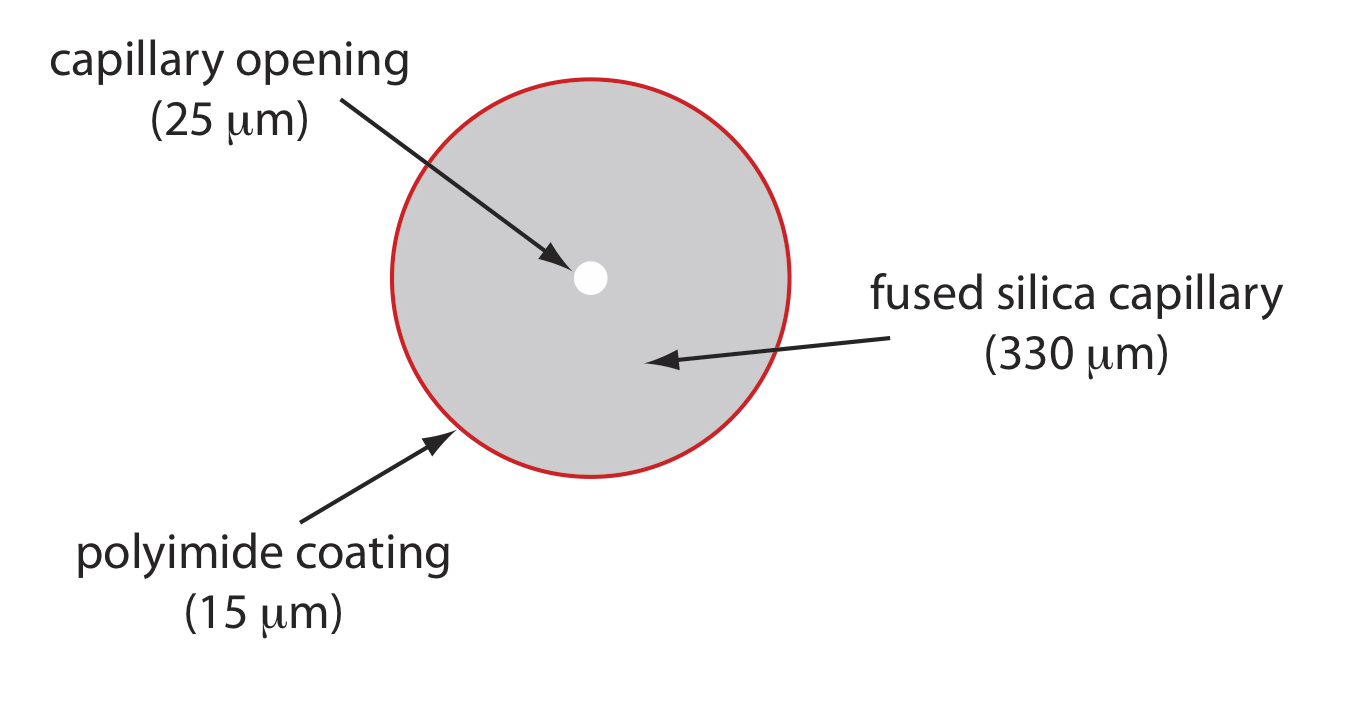
La estrecha apertura de la columna capilar y el grosor de sus paredes son importantes. Cuando se aplica un campo eléctrico a la solución tampón, la corriente fluye a través del capilar. Esta corriente conduce a la liberación de calor, que llamamos calefacción Joule. La cantidad de calor liberado es proporcional al radio del capilar y a la magnitud del campo eléctrico. El calentamiento de Joule es un problema porque cambia la viscosidad del tampón, siendo la solución en el centro del capilar menos viscosa que la que está cerca de las paredes capilares. Debido a que la movilidad electroforética de un soluto depende de su viscosidad (ver Ecuación\ ref {12.2}), las especies de solutos en el centro del capilar migran a una velocidad más rápida que las cercanas a las paredes capilares. El resultado es una fuente adicional de ensanchamiento de banda que degrada la separación. Los capilares con diámetros internos más pequeños generan menos calentamiento Joule, y los capilares con diámetros exteriores más grandes son más efectivos para disipar el calor. Colocar el tubo capilar dentro de una camisa termostática es otro método para minimizar el efecto del calentamiento Joule; en este caso un diámetro exterior más pequeño permite una disipación más rápida de la energía térmica.
Inyectar la muestra
Existen dos métodos comunes para inyectar una muestra en una columna de electroforesis capilar: la inyección hidrodinámica y la inyección electrocinética. En ambos métodos el tubo capilar se llena con la solución tampón. Un extremo del tubo capilar se coloca en el depósito de destino y el otro extremo se coloca en el vial de muestra.
La inyección hidrodinámica utiliza presión para forzar una pequeña porción de la muestra en el tubo capilar. Se aplica una diferencia de presión a través del capilar, ya sea presurizando el vial de muestra o aplicando vacío al depósito de destino. El volumen de muestra inyectada, en litros, viene dado por la siguiente ecuación
\[V_{\text {inj}}=\frac{\Delta P d^{4} \pi t}{128 \eta L} \times 10^{3} \label{12.12}\]
donde\(\Delta P\) está la diferencia de presión a través del capilar en pascales, d es el diámetro interno del capilar en metros, t es la cantidad de tiempo que se aplica la presión en segundos,\(\eta\) es la viscosidad del tampón en kg m —1 s —1, y L es la longitud del tubo capilar en metros. El factor de 10 3 cambia las unidades de metros cúbicos a litros.
Para una inyección hidrodinámica trasladamos el capilar desde el depósito fuente a la muestra. El ánodo permanece en el depósito fuente. También es posible una inyección hidrodinámica si elevamos el vial de muestra por encima del depósito de destino e insertamos brevemente el capilar lleno.
En una inyección hidrodinámica aplicamos una diferencia de presión de\(2.5 \times 10^3\) Pa (a\(\Delta P \approx 0.02 \text{ atm}\)) por 2 s a un tubo capilar de 75 cm de largo con un diámetro interno de 50 μm. Suponiendo que la viscosidad del tampón es de 10 —3 kg m —1 s —1, ¿qué volumen y longitud de muestra inyectamos?
Solución
Hacer sustituciones apropiadas en la Ecuación\ ref {12.12} da el volumen de la muestra como
\[V_{inj}=\frac{\left(2.5 \times 10^{3} \text{ kg} \text{ m}^{-1} \text{ s}^{-2}\right)\left(50 \times 10^{-6} \text{ m}\right)^{4}(3.14)(2 \text{ s})}{(128)\left(0.001 \text{ kg} \text{ m}^{-1} \text{ s}^{-1}\right)(0.75 \text{ m})} \times 10^{3} \mathrm{L} / \mathrm{m}^{3} \nonumber\]
\[V_{inj} = 1 \times 10^{-9} \text{ L} = 1 \text{ nL} \nonumber\]
Debido a que el interior del capilar es cilíndrico, la longitud de la muestra, l, es fácil de calcular usando la ecuación para el volumen de un cilindro; así
\[l=\frac{V_{\text {inj}}}{\pi r^{2}}=\frac{\left(1 \times 10^{-9} \text{ L}\right)\left(10^{-3} \text{ m}^{3} / \mathrm{L}\right)}{(3.14)\left(25 \times 10^{-6} \text{ m}\right)^{2}}=5 \times 10^{-4} \text{ m}=0.5 \text{ mm} \nonumber\]
Supongamos que necesita limitar su inyección a menos del 0.20% de la longitud del capilar. Usando la información del Ejemplo 12.7.1 , ¿cuál es el tiempo máximo de inyección para una inyección hidrodinámica?
- Responder
-
El capilar mide 75 cm de largo, lo que significa que 0.20% de la longitud máxima de esa muestra es de 0.15 cm. Para convertir esto al volumen máximo de muestra utilizamos la ecuación para el volumen de un cilindro.
\[V_{i n j}=l \pi r^{2}=(0.15 \text{ cm})(3.14)\left(25 \times 10^{-4} \text{ cm}\right)^{2}=2.94 \times 10^{-6} \text{ cm}^{3} \nonumber\]
Dado que 1 cm 3 es equivalente a 1 mL, el volumen máximo es\(2.94 \times 10^{-6}\) mL o\(2.94 \times 10^{-9}\) L. Para encontrar el tiempo máximo de inyección, primero resolvemos la Ecuación\ ref {12.12} para t
\[t=\frac{128 V_{inj} \eta L}{P d^{4} \pi} \times 10^{-3} \text{ m}^{3} / \mathrm{L} \nonumber\]
y luego hacer las sustituciones apropiadas.
\[t=\frac{(128)\left(2.94 \times 10^{-9} \text{ L}\right)\left(0.001 \text{ kg } \text{ m}^{-1} \text{ s}^{-1}\right)(0.75 \text{ m})}{\left(2.5 \times 10^{3} \text{ kg } \mathrm{m}^{-1} \text{ s}^{-2}\right)\left(50 \times 10^{-6} \text{ m}\right)^{4}(3.14)} \times \frac{10^{-3} \text{ m}^{3}}{\mathrm{L}} = 5.8 \text{ s} \nonumber\]
El tiempo máximo de inyección, por lo tanto, es de 5.8 s.
En una inyección electrocinética colocamos tanto el capilar como el ánodo en la muestra y aplicamos brevemente un potencial. El volumen de muestra inyectada es el producto del área de sección transversal del capilar y la longitud del capilar ocupado por la muestra. A su vez, esta longitud es el producto de la velocidad del soluto (ver Ecuación\ ref {12.6}) y el tiempo; así
\[V_{inj} = \pi r^2 L = \pi r^2 (\mu_{ep} + \mu_{eof})E^{\prime}t \label{12.13}\]
donde r es el radio del capilar, L es la longitud del capilar y\(E^{\prime}\) es el campo eléctrico efectivo en la muestra. Una consecuencia importante de la Ecuación\ ref {12.13} es que una inyección electrocinética está sesgada hacia solutos con mayores movilidades electroforéticas. Si dos solutos tienen concentraciones iguales en una muestra, inyectamos un volumen mayor y, por lo tanto, más moles, del soluto con el mayor\(\mu_{ep}\).
El campo eléctrico en la muestra es diferente que el campo eléctrico en el resto del capilar porque la muestra y el tampón tienen diferentes composiciones iónicas. En general, la fuerza iónica de la muestra es menor, lo que hace que su conductividad sea más pequeña. El campo eléctrico efectivo es
\[E^{\prime} = E \times \frac {\chi_\text{buffer}} {\chi_\text{sample}}\nonumber\]
donde\(\chi_\text{buffer}\) y\(\chi_{sample}\) son las conductividades del tampón y la muestra, respectivamente.
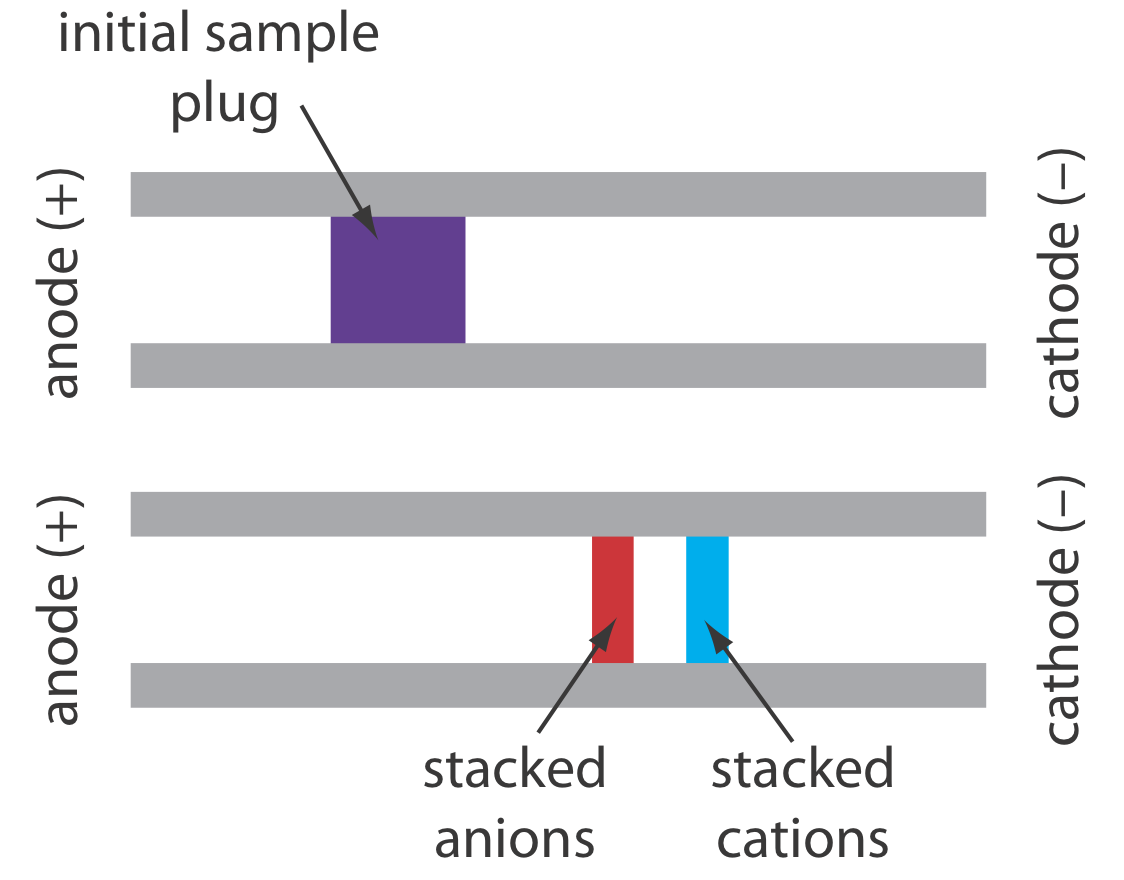
Cuando la concentración de un analito es demasiado pequeña para detectarlo de manera confiable, tal vez sea posible inyectarlo de una manera que aumente su concentración. Este método de inyección se llama apilamiento. El apilamiento se logra colocando la muestra en una solución cuya fuerza iónica es significativamente menor que la del tampón en el tubo capilar. Debido a que el tapón de muestra tiene una menor concentración de iones tampón, la intensidad de campo efectiva a través del tapón de muestra\(E^{\prime}\), es mayor que la del resto del capilar.
Sabemos por la Ecuación\ ref {12.1} que la velocidad electroforética es directamente proporcional al campo eléctrico. Como resultado, los cationes en el tapón de muestra migran hacia el cátodo con una mayor velocidad, y los aniones migran más lentamente, las especies neutras no se ven afectadas y se mueven con el flujo electroosmótico. Cuando los iones alcanzan sus respectivos límites entre el enchufe de muestra y el tampón, el campo eléctrico disminuye y la velocidad electroforética de los cationes disminuye y esa para los aniones aumenta. Como se muestra en la Figura 12.7.6 , el resultado es un apilamiento de cationes y aniones en zonas de muestreo separadas y más pequeñas. Con el tiempo, el tampón dentro del capilar se vuelve más homogéneo y la separación avanza sin apilamiento adicional.
Aplicando el Campo Eléctrico
La migración en electroforesis ocurre en respuesta a un campo eléctrico aplicado. La capacidad de aplicar un campo eléctrico grande es importante porque mayores voltajes conducen a tiempos de análisis más cortos (Ecuación\ ref {12.9}), separaciones más eficientes (Ecuación\ ref {12.10}) y mejor resolución (Ecuación\ ref {12.11}). Debido a que los tubos capilares perforados estrechos disipan el calentamiento Joule de manera tan eficiente, son posibles voltajes de hasta 40 kV.
Debido a los altos voltajes, asegúrese de seguir las pautas de seguridad de su instrumento.
Detectores
La mayoría de los detectores utilizados en HPLC también encuentran uso en electroforesis capilar. Entre los detectores más comunes se encuentran los basados en la absorción de radiación UV/Vis, fluorescencia, conductividad, amperometría y espectrometría de masas. Siempre que sea posible, la detección se realiza “en columna” antes de que los solutos se eluyan del tubo capilar y se produzca un ensanchamiento adicional de la banda.
Los detectores UV/Vis se encuentran entre los más populares. Debido a que la absorbancia es directamente proporcional a la longitud de la trayectoria, el pequeño diámetro del tubo capilar conduce a señales que son más pequeñas que las obtenidas en HPLC. Se han utilizado varios enfoques para aumentar la longitud de la ruta, incluyendo una celda de muestra en forma de Z y múltiples reflexiones (ver Figura 12.7.7 ). Los límites de detección son de aproximadamente 10 —7 M.
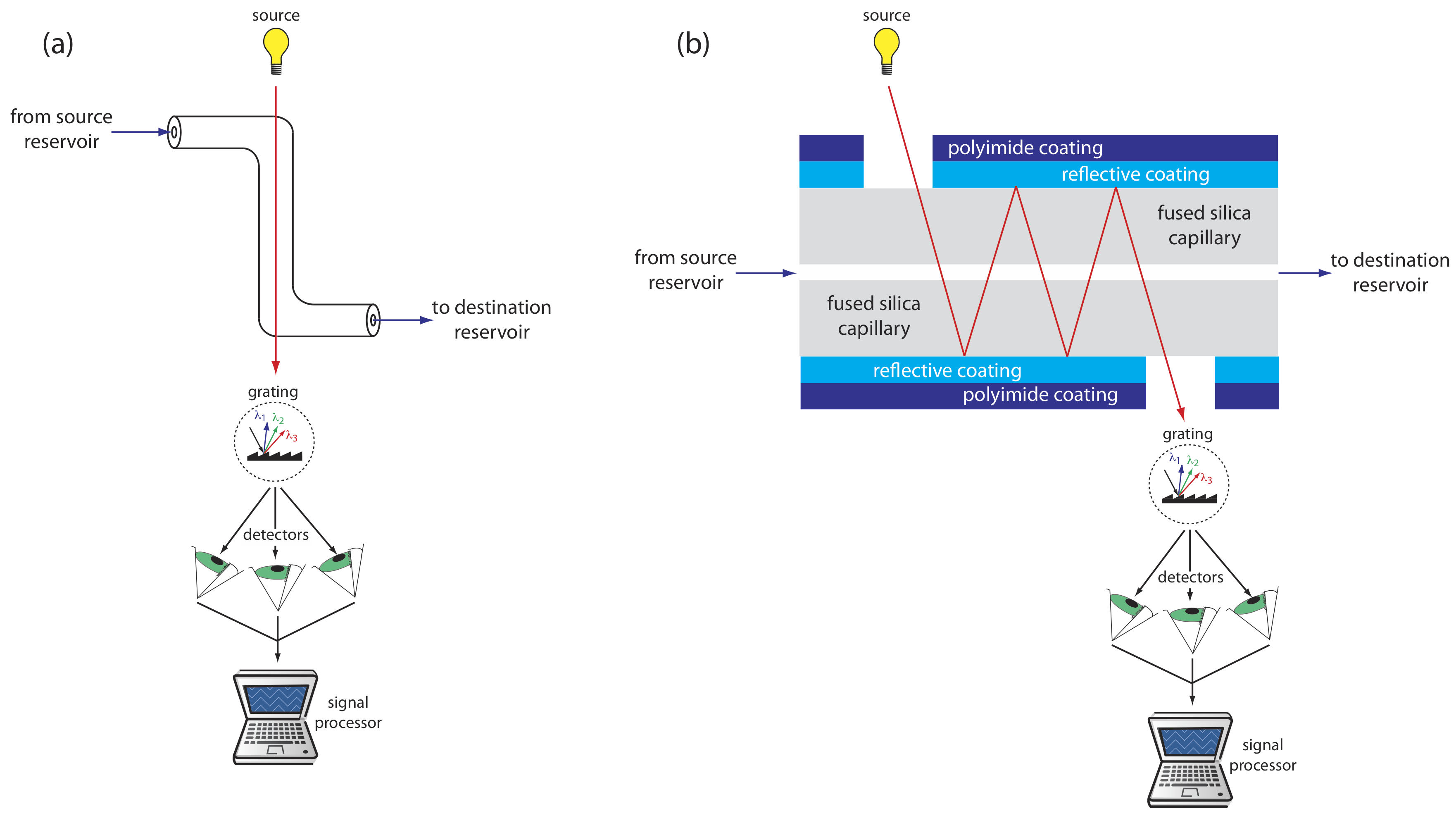
Se obtienen mejores límites de detección usando fluorescencia, particularmente cuando se usa un láser como fuente de excitación. Cuando se usa la detección de fluorescencia, se elimina una pequeña porción del recubrimiento protector del capilar y el rayo láser se enfoca en la porción interna del tubo capilar. La emisión se mide en un ángulo de 90 o con respecto al láser. Debido a que el láser proporciona una fuente intensa de radiación que puede enfocarse en un punto estrecho, los límites de detección son tan bajos como 10 —16 M.
Los solutos que no absorben radiación UV/Vis o que no experimentan fluorescencia pueden ser detectados por otros detectores. El cuadro 12.7.1 proporciona una lista de detectores para electroforesis capilar junto con algunas de sus características importantes.
Métodos de electroforesis capilar
Existen varias formas diferentes de electroforesis capilar, cada una de las cuales tiene sus ventajas particulares. Cuatro de estos métodos se describen brevemente en esta sección.
Electroforesis de Zona Capilar (CZE)
La forma más simple de electroforesis capilar es la electroforesis de zona capilar. En CZE llenamos el tubo capilar con un tampón y, después de cargar la muestra, colocamos los extremos del tubo capilar en depósitos que contienen tampón adicional. Por lo general, el extremo del capilar que contiene la muestra es el ánodo y los solutos migran hacia el cátodo a una velocidad determinada por sus respectivas movilidades electroforéticas y el flujo electroosmótico. Los cationes eluyen primero, con cationes más pequeños y con mayor carga eluyendo antes que los cationes más grandes con cargas más pequeñas. Las especies neutras eluyen como una sola banda. Los aniones son la última especie en eluir, siendo los aniones más pequeños y con mayor carga negativa los últimos en eluir.
Podemos invertir la dirección del flujo electroosmótico agregando una sal de alquilamonio a la solución tampón. Como se muestra en la Figura 12.7.8 , el extremo cargado positivamente de los iones alquilamonio se une a los iones silanato cargados negativamente en las paredes del capilar. La cola del ion alquilamonio es hidrofóbica y se asocia con la cola de otro ion alquilamonio. El resultado es una capa de cargas positivas que atraen aniones en el búfer. La migración de estos aniones solvatados hacia el ánodo invierte la dirección del flujo electroosmótico. El orden de elución es exactamente opuesto al observado en condiciones normales.
El recubrimiento de las paredes del capilar con un reactivo no iónico elimina el flujo electroosmótico. En esta forma de CZE los cationes migran del ánodo al cátodo. Los aniones eluyen en el reservorio fuente y las especies neutras permanecen estacionarias.
La electroforesis de zona capilar proporciona separaciones efectivas de especies cargadas, incluyendo aniones y cationes inorgánicos, ácidos orgánicos y aminas, y biomoléculas grandes como proteínas. Por ejemplo, se utilizó CZE para separar una mezcla de 36 iones inorgánicos y orgánicos en menos de tres minutos [Jones, W. R.; Jandik, P. J. Chromatog. 1992, 608, 385—393]. Una mezcla de especies neutras, por supuesto, no se puede resolver.
Cromatografía Micelar Electrocinética Capilar (MEKC)
Una limitación a la CZE es su incapacidad para separar especies neutras. La cromatografía capilar electrocinética micelar supera esta limitación al agregar un surfactante, como el dodecilsulfato de sodio (Figura 12.7.9 a) a la solución tampón. El dodecilsulfato de sodio, o SDS, consiste en una cola hidrófoba de cadena larga y un grupo funcional iónico cargado negativamente en su cabeza. Cuando la concentración de SDS es suficientemente grande se forma una micela. Una micela consiste en una aglomeración esférica de 40-100 moléculas de surfactante en la que las colas de hidrocarburos apuntan hacia adentro y las cabezas cargadas negativamente apuntan hacia afuera (Figura 12.7.9 b).
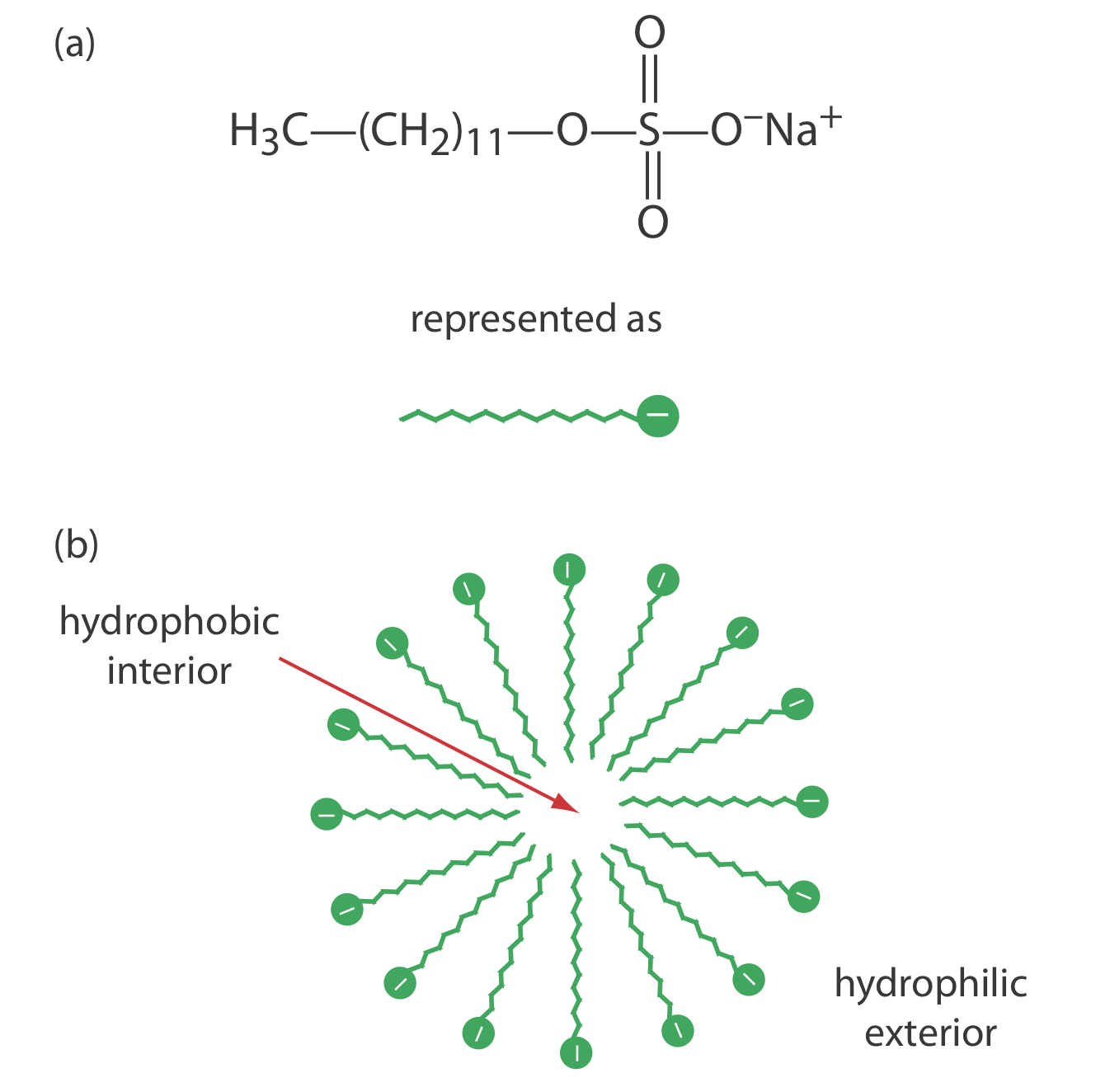
Debido a que las micelas tienen una carga negativa, migran hacia el cátodo con una velocidad menor que la velocidad de flujo electroosmótico. Las especies neutras se reparten entre las micelas y la solución tampón de manera similar a la partición de solutos entre las dos fases líquidas en HPLC. Debido a que existe una partición entre dos fases, incluimos el término descriptivo cromatografía en el nombre de las técnicas. Tenga en cuenta que en MEKC ambas fases son móviles.
El orden de elución de las especies neutras en MEKC depende de la medida en que cada especie se reparte en las micelas. Los neutros hidrofílicos son insolubles en el ambiente interno hidrofóbico de la micela y eluyen como una sola banda, como lo harían en CZE. Los solutos neutros que son extremadamente hidrofóbicos son completamente solubles en la micela, eluyendo con las micelas como una sola banda. Aquellas especies neutras que existen en un equilibrio de partición entre el tampón y las micelas eluyen entre las especies neutras completamente hidrófilas y completamente hidrofóbicas. Aquellas especies neutras que favorecen el tampón eluyen antes que las que favorecen las micelas. La cromatografía electrocinética micelar se utiliza para separar una amplia variedad de muestras, incluyendo mezclas de compuestos farmacéuticos, vitaminas y explosivos.
Electroforesis Capilar en Gel (CGE)
En la electroforesis capilar en gel el tubo capilar se rellena con un gel polimérico. Debido a que el gel es poroso, un soluto migra a través del gel con una velocidad determinada tanto por su movilidad electroforética como por su tamaño. La capacidad de efectuar una separación usando el tamaño es útil cuando los solutos tienen movilidades electroforéticas similares. Por ejemplo, los fragmentos de ADN de longitud variable tienen relaciones de carga a tamaño similares, lo que dificulta su separación por CZE. Debido a que los fragmentos de ADN son de diferente tamaño, es posible una separación de CGE.
El capilar utilizado para CGE generalmente se trata para eliminar el flujo electroosmótico para evitar que el gel se extruya desde el tubo capilar. Las muestras se inyectan electrocinéticamente porque el gel proporciona demasiada resistencia para el muestreo hidrodinámico. La aplicación primaria de CGE es la separación de biomoléculas grandes, incluyendo fragmentos de ADN, proteínas y oligonucleótidos.
Electrocromatografía Capilar (CEC)
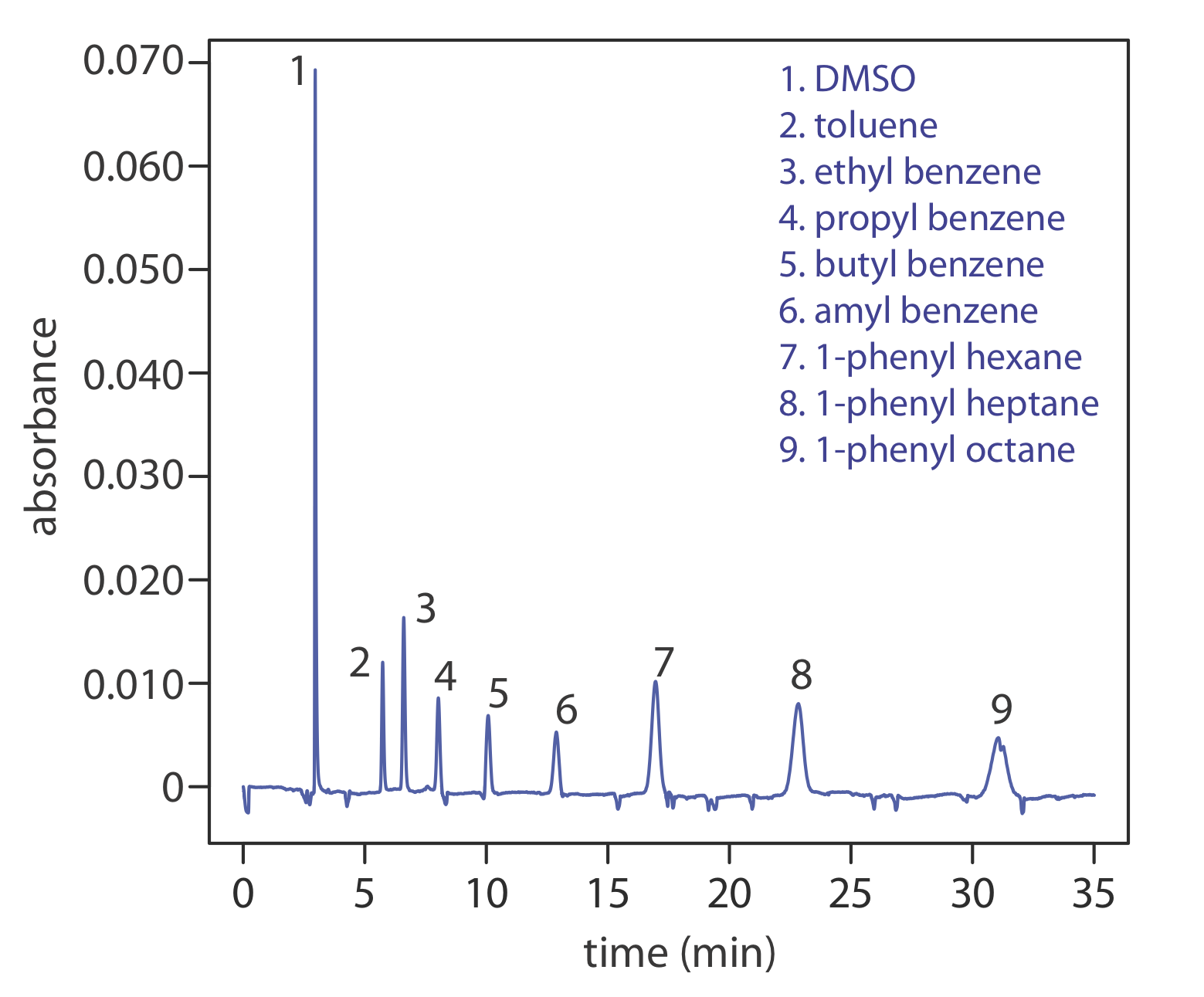
Otro enfoque para separar especies neutras es la electrocromatografía capilar. En CEC, el tubo capilar está empaquetado con partículas de 1.5—3 μm recubiertas con una fase estacionaria unida. Las especies neutras se separan en función de su capacidad de partición entre la fase estacionaria y el tampón, que se mueve como resultado del flujo electroosmótico; la Figura 12.7.10 proporciona un ejemplo representativo para la separación de una mezcla de hidrocarburos. Una separación CEC es similar a la separación por HPLC análoga, pero sin necesidad de bombas de alta presión. La eficiencia en CEC es mejor que en HPLC, y los tiempos de análisis son más cortos.
La mejor manera de apreciar los detalles teóricos y prácticos discutidos en esta sección es examinar cuidadosamente un método analítico típico. Aunque cada método es único, la siguiente descripción de la determinación de un complejo de vitamina B por electroforesis de zona capilar o por cromatografía capilar electrocinética micelar proporciona un ejemplo instructivo de un procedimiento típico. La descripción aquí se basa en Smyth, W. F. Analytical Chem istry of Complex Matrices, Wiley Teubner: Chichester, Inglaterra, 1996, pp. 154—156.
Método Representativo 12.7.1: Determinación del Complejo de Vitamina B por CZE o MEKC
Descripción del método
Las vitaminas solubles en agua B 1 (clorhidrato de tiamina), B 2 (riboflavina), B 3 (niacinamida) y B 6 (clorhidrato de piridoxina) se determinan por CZE usando un tampón de tetraborato de sodihidrogenofosfato de sodio pH 9, o por MEKC usando el mismo tampón con el adición de dodecilsulfato de sodio. La detección es por absorción UV a 200 nm. Se utiliza un estándar interno de o-etoxibenzamida para estandarizar el método.
Procedimiento
Triturar una tableta de complejo de vitamina B y colocarla en un vaso de precipitados con 20.00 mL de una solución de metanol al 50% v/v que es 20 mM en tetraborato de sodio y 100.0 ppm en o-etoxibenzamida. Después de mezclar durante 2 min para asegurar que las vitaminas B se disuelven, pase una porción de 5.00-mL a través de un filtro de 0.45-μm para eliminar los aglutinantes insolubles. Cargar una muestra de aproximadamente 4 nL en una columna capilar con un diámetro interno de 50 μm. Para CZE la columna capilar contiene un tampón de tetraborato sódico-fosfato de sodio 20 mM pH 9. Para MEKC el tampón también es de 150 mM en dodecilsulfato de sodio. Aplicar un campo eléctrico de 40 kV/m para efectuar las separaciones CZE y MEKC.
Preguntas
1. El metanol, que eluye a los 4.69 min, se incluye como especie neutra para indicar el flujo electroosmótico. Al usar soluciones estándar de cada vitamina, los picos de CZE se encuentran a los 3.41 min, 4.69 min, 6.31 min y 8.31 min. Examine las estructuras y p K una información en la Figura 12.7.11 e identifique el orden en que eluyen las cuatro vitaminas B.
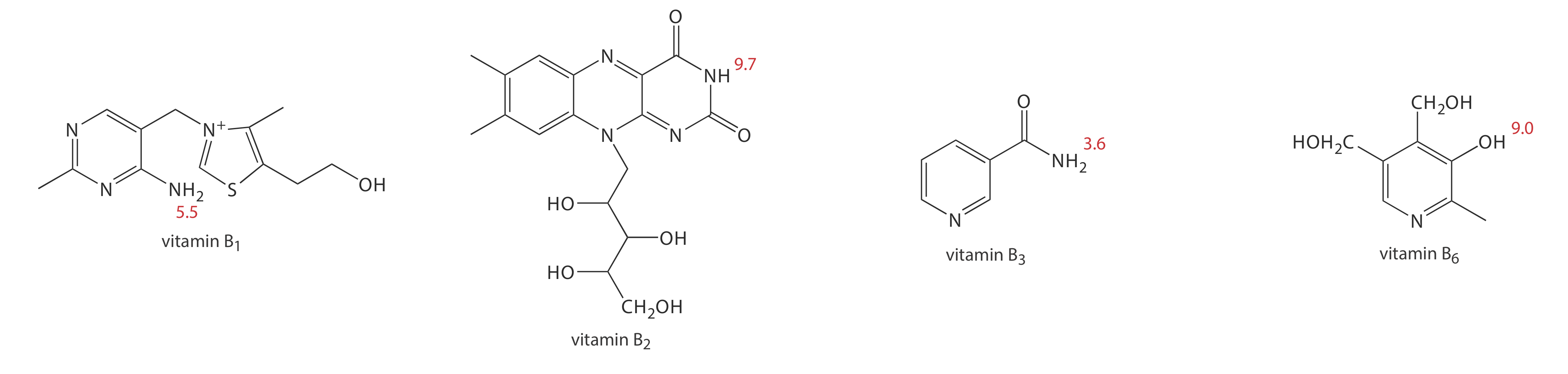
A un pH de 9, la vitamina B 1 es un catión y eluye antes que la especie neutra metanol; así es el compuesto que eluye a los 3.41 min. La vitamina B 3 es una especie neutra a pH de 9 y eluye con metanol a los 4.69 min. Las dos vitaminas B restantes son ácidos débiles que ionizan parcialmente a aniones de base débil a un pH de 9. De los dos, la vitamina B 6 es el ácido más fuerte (una p K a de 9.0 frente a una p K a de 9.7) y está presente en mayor medida en su forma aniónica. La vitamina B 6, por lo tanto, es la última de las vitaminas en eluir.
2. El orden de elución al usar MEKC es vitamina B 3 (5.58 min), vitamina B 6 (6.59 min), vitamina B 2 (8.81 min) y vitamina B 1 (11.21 min). ¿Qué conclusiones puedes sacar sobre la solubilidad de las vitaminas B en las micelas de dodecilsulfato de sodio? Las micelas eluyen a 17.7 min.
El tiempo de elución para la vitamina B 1 muestra el mayor cambio, aumentando de 3.41 min a 11.21 minutos. Claramente, la vitamina B 1 tiene la mayor solubilidad en las micelas. La vitamina B 2 y la vitamina B3 tienen una solubilidad más limitada en las micelas, y muestran tiempos de elución ligeramente más largos en presencia de las micelas. Curiosamente, el tiempo de elución de la vitamina B 6 disminuye en presencia de las micelas.
3. Para el trabajo cuantitativo se agrega un estándar interno de o-etoxibenzamida a todas las muestras y estándares. ¿Por qué es necesario un estándar interno?
Aunque no se especifica el método de inyección, ni una inyección hidrodinámica ni una inyección electrocinética son particularmente reproducibles. El uso de una norma interna compensa esta limitación.
Evaluación
Cuando se compara con GC y HPLC, la electroforesis capilar proporciona niveles similares de precisión, precisión y sensibilidad, y proporciona un grado comparable de selectividad. La cantidad de material inyectado en una columna electroforética capilar es significativamente menor que la de GC y HPLC, típicamente 1 nL versus 0.1 μL para GC capilar y 1—100 μL para HPLC. Sin embargo, los límites de detección para la electroforesis capilar son 100—1000 veces más pobres que los de GC y HPLC. Las ventajas más significativas de la electroforesis capilar son las mejoras en la eficiencia de separación, el tiempo y el costo. Las columnas electroforéticas capilares contienen sustancialmente más placas teóricas (\(\approx 10^6\)placas/m) que las que se encuentran en las columnas HPLC (\(\approx 10^5\)placas/m) y GC capilar (\(\approx 10^3\)placas/m), proporcionando una resolución y una capacidad máxima inigualables. Las separaciones en electroforesis capilar son rápidas y eficientes. Además, el pequeño volumen de la columna capilar significa que una separación por electroforesis capilar requiere solo unos pocos microlitros de tampón, en comparación con 20—30 ml de fase móvil para una separación típica de HPLC.