
12.8: Problemas
- Page ID
- 75667

1. Se obtuvieron los siguientes datos para cuatro compuestos separados en una columna capilar de 20 m.
| compuesto | t r (min) | w (min) |
|---|---|---|
| A | 8.04 | 0.15 |
| B | 8.26 | 0.15 |
| C | 8.43 | 0.16 |
a) Calcular el número de placas teóricas para cada compuesto y el número promedio de placas teóricas para la columna, en mm.
(b) Calcular la altura promedio de una placa teórica.
c) Explicar por qué es posible que cada compuesto tenga un número diferente de placas teóricas.
2. Utilizando los datos del Problema 1, se calculan los factores de resolución y selectividad para cada par de compuestos adyacentes. Para la resolución, use tanto la ecuación 12.2.1 como la ecuación 12.3.3, y compare sus resultados. Discuta cómo podría mejorar la resolución entre los compuestos B y C. El tiempo de retención para un soluto no retenido es de 1.19 min.
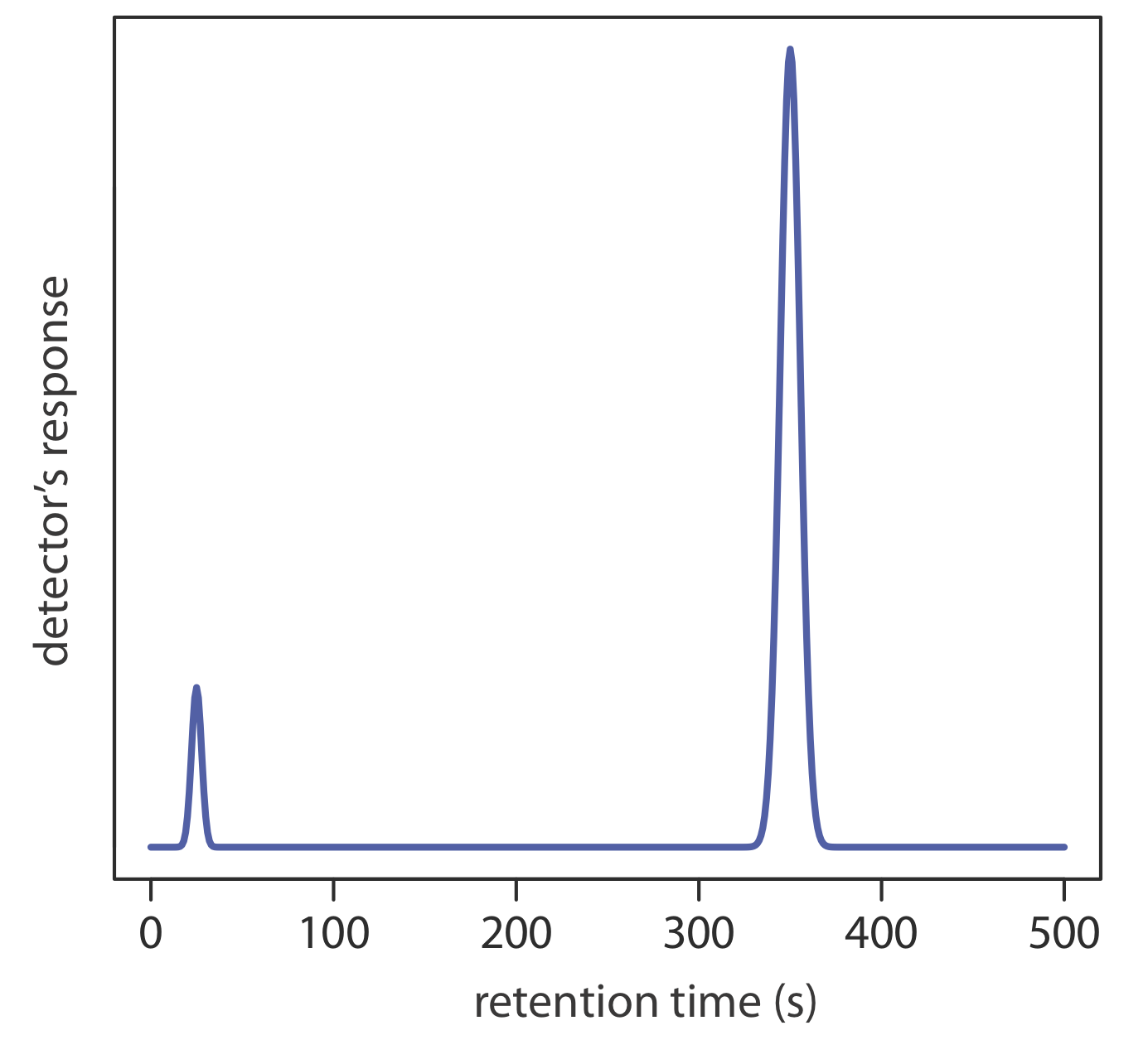
3. Utilice el cromatograma de la Figura 12.8.1 , obtenido usando una columna de 2 m, para determinar los valores para t r, w\(t_r^{\prime}\),, k, N y H.
4. Utilice el cromatograma parcial de la Figura 12.8.2 para determinar la resolución entre las dos bandas de solutos.
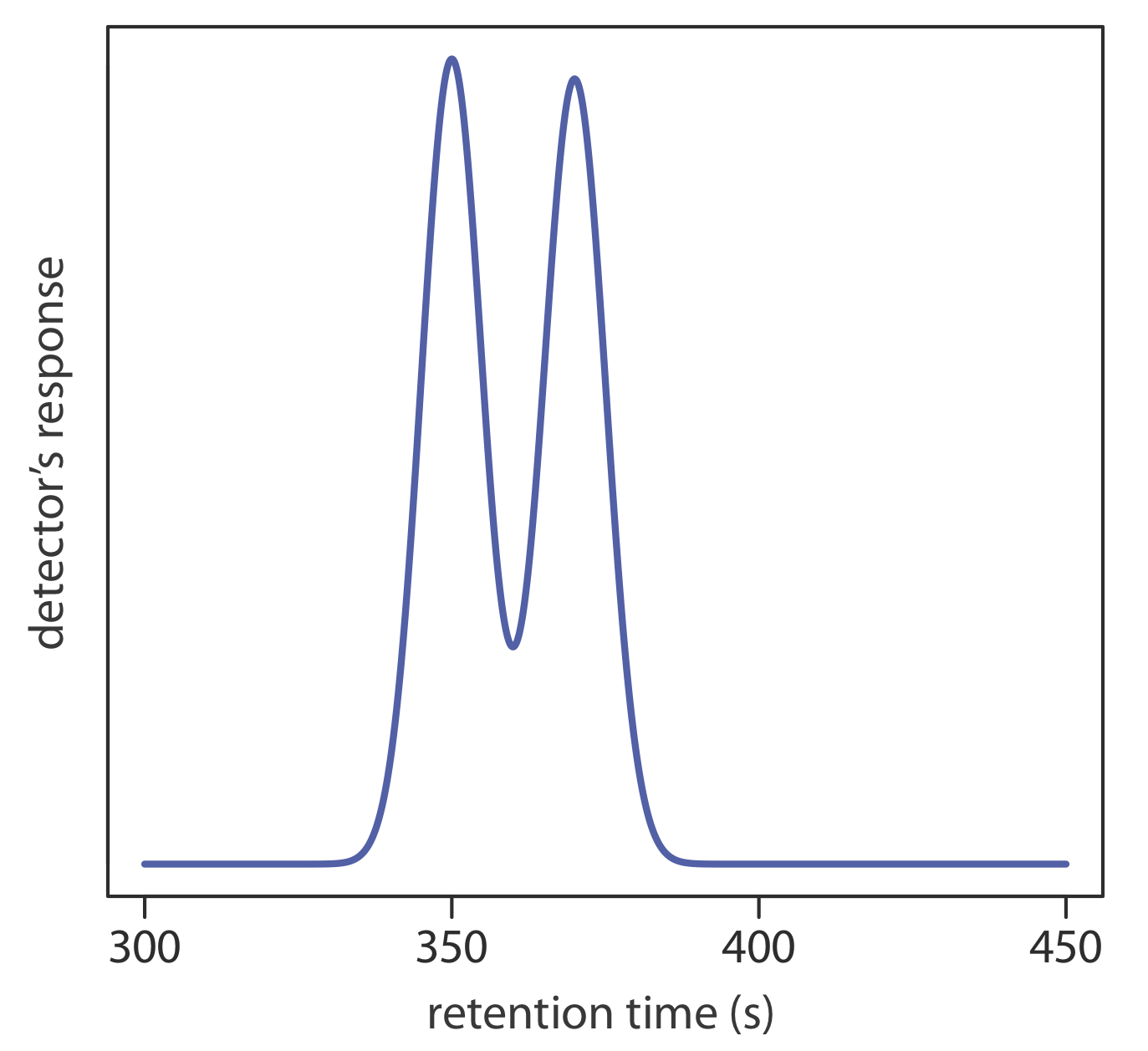
5. El cromatograma en el Problema 4 se obtuvo en una columna de 2 m con un tiempo muerto de columna de 50 s. Supongamos que desea aumentar la resolución entre los dos componentes a 1.5. Sin cambiar la altura de una placa teórica, ¿qué longitud de columna necesitas? ¿Qué altura de una placa teórica necesitas para lograr una resolución de 1.5 sin aumentar la longitud de la columna?
6. Complete la siguiente tabla.
| N B | \(\alpha\) | k B | R |
|---|---|---|---|
| 100000 | \ (\ alfa\) ">1.05 | 0.50 | |
| 10000 | \ (\ alfa\) ">1.10 | 1.50 | |
| 10000 | \ (\ alfa\) "> | 4.0 | 1.00 |
| \ (\ alfa\) ">1.05 | 3.0 | 1.75 |
7. Moody estudió la eficiencia de una separación GC de 2-butanona en una columna empaquetada con ftalato de dinonilo [Moody, H. W. J. Chem. Educ. 1982, 59, 218—219]. Al evaluar la altura de la placa en función del caudal se obtuvo una ecuación de van Deemter para la cual A es 1.65 mm, B es 25.8 mm•mL min —1 y C es 0.0236 mm•min mL —1.
(a) Preparar una gráfica de H versus u para caudales entre 5 —120 mL/min.
b) ¿Para qué rango de caudales tiene cada término de la ecuación de Van Deemter el mayor efecto?
c) ¿Cuál es el caudal óptimo y la altura correspondiente de una placa teórica?
d) Para las columnas tubulares abiertas ya no se necesita el término A. Si los términos B y C permanecen sin cambios, ¿cuál es el caudal óptimo y la altura correspondiente de una placa teórica?
e) En comparación con la columna empaquetada, ¿cuántas placas teóricas más hay en la columna tubular abierta?
8. Hsieh y Jorgenson prepararon columnas de HPLC de 12—33 μm de diámetro interno empacadas con partículas esféricas de 5.44-μm en fase estacionaria [Hsieh, S.; Jorgenson, J. W. Anal. Chem. 1996, 68, 1212—1217]. Para evaluar estas columnas midieron la altura reducida de la placa, h, en función del caudal reducido, v,
\[b=\frac{H}{d_{p}} \quad v=\frac{u d_{p}}{D_{m}} \nonumber\]
donde d p es el diámetro de partícula y D m es el coeficiente de difusión del soluto en la fase móvil. Los datos se analizaron mediante parcelas van Deemter. La siguiente tabla contiene una porción de sus resultados para la norepinefrina.
| diámetro interno (µm) | A | B | C |
|---|---|---|---|
| 33 | 0.63 | 1.32 | 0.10 |
| 33 | 0.67 | 1.30 | 0.08 |
| 23 | 0.40 | 1.34 | 0.09 |
| 23 | 0.58 | 1.11 | 0.09 |
| 17 | 0.31 | 1.47 | 0.11 |
| 17 | 0.40 | 1.41 | 0.11 |
| 12 | 0.22 | 1.53 | 0.11 |
| 12 | 0.19 | 1.27 | 0.12 |
(a) Construya parcelas separadas de Van Deemter utilizando los datos de la primera fila y en la última fila para caudales reducidos en el rango de 0.7—15. Determinar el caudal óptimo y la altura de placa para cada caso dado d p = 5.44 μm y D m =\(6.23 \times 10^{-6}\) cm 2 s —1.
(b) El término A en la ecuación de van Deemter está fuertemente correlacionado con el diámetro interno de la columna, con columnas de menor diámetro que proporcionan valores más pequeños de A. Ofrezca una explicación para esta observación. Consejo: considere cuántas partículas pueden caber a través de un capilar de cada diámetro.
Al comparar columnas, los cromatógrafos suelen utilizar parámetros reducidos y adimensionales. Al incluir el tamaño de partícula y el coeficiente de difusión del soluto, la altura reducida de la placa y el caudal reducido corrigen las diferencias entre el material de empaque, el soluto y la fase móvil.
9. Una mezcla de n-heptano, tetrahidrofurano, 2-butanona y n-propanol eluye en este orden cuando se usa una fase estacionaria polar como Carbowax. El orden de elución es exactamente el opuesto cuando se usa una fase estacionaria no polar tal como polidimetilsiloxano. Explicar el orden de elución en cada caso.
10. El análisis de trihalometanos en agua potable se describe en el Método Representativo 12.4.1. Un único estándar que contiene los cuatro trihalometanos da los siguientes resultados.
| compuesto | concentración (ppb) | área de pico |
|---|---|---|
| CHCl 3 | 1.30 | \(1.35 \times 10^4\) |
| ChCl 2 Br | 0.90 | \(6.12 \times 10^4\) |
| CHCLBr 2 | 4.00 | \(1.71 \times 10^4\) |
| CHBr 3 | 1.20 | \(1.52 \times 10^4\) |
El análisis del agua recolectada de un bebedero da áreas de\(1.56 \times 10^4\)\(5.13 \times10^4\),\(1.49 \times 10^4\),, y\(1.76 \times 10^4\) para, respectivamente, CHCl 3, CHCl 2 Br, CHClBr 2 y ChBr 3. Todas las áreas de los picos se corrigieron por variaciones en los volúmenes de inyección usando un estándar interno de 1,2-dibromopentano. Determinar la concentración de cada uno de los trihalometanos en la muestra de agua.
11. Zhou y sus colegas determinaron el% w/w H 2 O en metanol por columna capilar GC utilizando una fase estacionaria no polar y un detector de conductividad térmica [Zhou, X.; Hines, P. A.; White, K. C.; Borer, M. W. Anal. Chem. 1998, 70, 390—394]. Una serie de estándares de calibración dieron los siguientes resultados.
| % p/p H 2 O | altura de pico (unidades arb.) |
|---|---|
| 0.00 | 1.15 |
| 0.0145 | 2.74 |
| 0.0472 | 6.33 |
| 0.0951 | 11.58 |
| 0.1757 | 20.43 |
| 0.2901 | 32.97 |
(a) ¿Cuál es el% w/w H 2 O en una muestra que tiene una altura máxima de 8.63?
(b) El% p/p de H 2 O en un antibiótico liofilizado se determina de la siguiente manera. Se coloca una muestra de 0.175 g en un vial junto con 4.489 g de metanol. El agua en el vial se extrae en el metanol. El análisis de la muestra dio una altura máxima de 13.66. ¿Cuál es el% w/w H 2 O en el antibiótico?
12. Loconto y compañeros de trabajo describen un método para determinar los niveles traza de agua en el suelo [Loconto, P. R.; Pan, Y. L.; Voice, T. C. LC•GC 1996, 14, 128—132]. El método aprovecha la reacción del agua con carburo de calcio, CaC 2, para producir gas acetileno, C 2 H 2. Al llevar a cabo la reacción en un vial sellado, se determina la cantidad de acetileno producido muestreando el espacio de cabeza. En un análisis típico se coloca una muestra de suelo en un vial sellado con CaC 2. El análisis del espacio de cabeza da una señal corregida en blanco de\(2.70 \times 10^5\). Se prepara una segunda muestra de la misma manera excepto que se agrega una adición estándar de 5.0 mg H 2 O/g de suelo, dando una señal corregida en blanco de\(1.06 \times 10^6\). Determinar los miligramos H 2 O/g de suelo en la muestra de suelo.
13. Van Atta y Van Atta utilizaron cromatografía de gases para determinar el salicilato de metilo% v/v en alcohol para frotar [Van Atta, R. E.; Van Atta, R. L. J. Chem. Educ. 1980, 57, 230—231]. Se preparó un conjunto de adiciones estándar transfiriendo 20.00 mL de alcohol para frotar para separar matraces volumétricos de 25 mL y pipetear 0.00 mL, 0.20 mL y 0.50 mL de salicilato de metilo a los matraces. Los tres matraces se diluyeron a volumen usando isopropanol. El análisis de las tres muestras dio alturas de pico para salicilato de metilo de 57.00 mm, 88.5 mm y 132.5 mm, respectivamente. Determinar el salicilato de metilo% v/v en el alcohol para frotar.
14. La cantidad de alcanfor en una pomada analgésica se determina por GC utilizando el método de estándares internos [Pant, S. K.; Gupta, P. N.; Thomas, K. M.; Maitin, B. K.; Jain, C. L. LC•GC 1990, 8, 322—325]. Se prepara una muestra estándar colocando 45.2 mg de alcanfor y 2.00 mL de una solución patrón interno 6.00 mg/mL de hidrato de terpeno en un matraz aforado de 25 mL y diluyendo a volumen con CCl 4. Cuando se inyecta una muestra de aproximadamente 2 μL del estándar, las señales FID para los dos componentes se miden (en unidades arbitrarias) como 67.3 para alcanfor y 19.8 para hidrato de terpeno. Se prepara una muestra de 53.6 mg de una pomada analgésica para su análisis colocándola en un matraz Erlenmeyer de 50 mL junto con 10 mL de CCl 4. Después de calentar a 50 o C en un baño de agua, la muestra se enfría por debajo de la temperatura ambiente y se filtra. El residuo se lava con dos porciones de 5 mL de CCl 4 y los filtrados combinados se recogen en un matraz aforado de 25 mL. Después de agregar 2.00 mL de la solución patrón interno, el contenido del matraz se diluye a volumen con CCl 4. El análisis de una muestra de aproximadamente 2 μL da señales FID de 13.5 para el hidrato de terpeno y 24.9 para el alcanfor. Reporte el %w/w de alcanfor en la pomada analgésica.
15. La concentración de residuos de plaguicidas en productos agrícolas, como las naranjas, se determina por CG-MS [Feigel, C. Varian GC/MS Application Note, Número 52]. Los residuos de plaguicidas se extraen de la muestra usando cloruro de metileno y se concentran evaporando el cloruro de metileno a un volumen menor. La calibración se realiza usando antraceno-d 10 como estándar interno. En un estudio para determinar las partes por mil millones de epóxido de heptacloro en naranjas, se corta una muestra de 50.0-g de corteza de naranja y se extrae con 50.00 mL de cloruro de metileno. Después de eliminar cualquier material insoluble por filtración, el cloruro de metileno se reduce en volumen, se agrega una cantidad conocida del patrón interno y se diluye a 10 mL en un matraz aforado. El análisis de la muestra da una relación pico-área (A analito/A intstd) de 0.108. Una serie de patrones de calibración, cada uno conteniendo la misma cantidad de antraceno-d 10 que la muestra, da los siguientes resultados.
| ppb epóxido de heptacloro | Un analito/A intstd |
|---|---|
| 20.0 | 0.065 |
| 60.0 | 0.153 |
| 200.0 | 0.637 |
| 500.0 | 1.554 |
| 1000.0 | 3.198 |
Reporte los nanogramos por gramo de residuo de epóxido de heptacloro en las naranjas.
16. Los tiempos de retención ajustados para octano, tolueno y nonano en una columna GC particular son 15.98 min, 17.73 min y 20.42 min, respectivamente. ¿Cuál es el índice de retención para cada compuesto?
17. Se recolectaron los siguientes datos para una serie de alcanos normales utilizando una fase estacionaria de Carbowax 20M.
| alcano | \(t_r^{\prime}\)(min) |
|---|---|
| pentano | \ (t_r^ {\ prime}\) (min) ">0.79 |
| hexano | \ (t_r^ {\ prime}\) (min) ">1.99 |
| heptano | \ (t_r^ {\ prime}\) (min) ">4.47 |
| octano | \ (t_r^ {\ prime}\) (min) ">14.12 |
| nonano | \ (t_r^ {\ prime}\) (min) ">33.11 |
¿Cuál es el índice de retención para un compuesto cuyo tiempo de retención ajustado es de 9.36 min?
18. Se reportaron los siguientes datos para el análisis cromatográfico de gases de p-xileno y metilisobutilcetona (MIBK) en una columna capilar [Marriott, P. J.; Carpenter, P. D. J. Chem. Educ. 1996, 73, 96—99].
| modo de inyección | compuesto | t r (min) | área de pico (unidades arb.) | ancho de pico (min) |
|---|---|---|---|---|
| split | MIBK | 1.878 | 54285 | 0.028 |
| p-xileno | 5.234 | 123483 | 0.044 | |
| sin divisiones | MIBK | 3.420 | 2493005 | 1.057 |
| p-xileno | 5.795 | 3396656 | 1.051 |
Explique la diferencia en los tiempos de retención, las áreas de los picos y los anchos de los picos al cambiar de una inyección dividida a una inyección sin división.
19. Otto y Wegscheider reportan los siguientes factores de retención para la separación en fase inversa del ácido 2-aminbenzoico en una columna C 18 al usar metanol al 10% v/v como fase móvil [Otto, M.; Wegscheider, W. J. Chromatog. 1983, 258, 11—22].
| pH | k |
|---|---|
| 2.0 | 10.5 |
| 3.0 | 16.7 |
| 4.0 | 15.8 |
| 5.0 | 8.0 |
| 6.0 | 2.2 |
| 7.0 | 1.8 |
Explicar el efecto del pH sobre el factor de retención para 2-aminobenceno.
20. Haddad y asociados reportan los siguientes factores de retención para la separación en fase inversa de salicilamida y cafeína [Haddad, P.; Hutchins, S.; Tuffy, M. J. Chem. Educ. 1983, 60, 166-168].
| % v/v de metanol | 30% | 35% | 40% | 45% | 50% | 55% |
| k sal | 2.4 | 1.6 | 1.6 | 1.0 | 0.7 | 0.7 |
| k caff | 4.3 | 2.8 | 2.3 | 1.4 | 1.1 | 0.9 |
a) Explicar las tendencias en los factores de retención de estos compuestos.
(b) ¿Cuál es la ventaja de usar una fase móvil con un% v/v de metanol más pequeño? ¿Hay alguna desventaja?
21. Supongamos que necesitas separar una mezcla de ácido benzoico, aspartamo y cafeína en un refresco dietético. La siguiente información está disponible.
| t r en fase móvil acuosa de pH | ||||
| compuesto | 3.0 | 3.5 | 4.0 | 4.5 |
| ácido benzoico | 7.4 | 7.0 | 6.9 | 4.4 |
| aspartamo | 5.9 | 6.0 | 7.1 | 8.1 |
| cafeína | 3.6 | 3.7 | 4.1 | 4.4 |
(a) Explicar el cambio en el tiempo de retención de cada compuesto.
(b) Preparar una gráfica única que muestre el tiempo de retención versus pH para cada compuesto. Usando su parcela, identifique un nivel de pH que produzca una separación aceptable.
22. La composición de un comprimido multivitamínico se determina mediante HPLC con un detector UV/Vis de matriz de diodos. Se inyecta en la HPLC una muestra estándar de 5 μL que contiene 170 ppm de vitamina C, 130 ppm de niacina, 120 ppm de niacinamida, 150 ppm de piridoxina, 60 ppm de tiamina, 15 ppm de ácido fólico y 10 ppm de riboflavina, dando señales (en unidades arbitrarias) de, respectivamente, 0.22, 1.35, 0.90, 1.37, 0.82, 0.36 y 0.29. El comprimido multivitamínico se prepara para su análisis moliendo en polvo y transfiriéndolo a un matraz Erlenmeyer de 125 ml que contiene 10 mL de NH 3 al 1% v/v en dimetilsulfóxido. Después de sonicación en un baño ultrasónico durante 2 min, se agregan 90 mL de ácido acético al 2% y la mezcla se agita durante 1 min y se sonica a 40 o C durante 5 min. Luego, el extracto se filtra a través de un filtro de membrana de 0.45-μm. La inyección de una muestra de 5 μL en la HPLC da señales de 0.87 para vitamina C, 0.00 para niacina, 1.40 para niacinamida, 0.22 para piridoxina, 0.19 para tiamina, 0.11 para ácido fólico y 0.44 para riboflavina. Reporte los miligramos de cada vitamina presente en la tableta.
23. La cantidad de cafeína en un comprimido analgésico se determinó por HPLC usando una curva de calibración normal. Se prepararon soluciones estándar de cafeína y se analizaron mediante un bucle de inyección de volumen fijo de 10-μL. Los resultados para los estándares se resumen en la siguiente tabla.
| concentración (ppm) | señal (unidades arb.) |
|---|---|
| 50.0 | 8.354 |
| 100.0 | 16925 |
| 150.0 | 25218 |
| 200.0 | 33584 |
| 250.0 | 42002 |
La muestra se prepara colocando una sola tableta analgésica en un vaso pequeño y añadiendo 10 mL de metanol. Después de dejar que la muestra se disuelva, el contenido del vaso de precipitados, incluido el aglutinante insoluble, se transfiere cuantitativamente a un matraz aforado de 25 ml y se diluye a volumen con metanol. Luego se filtra la muestra y se transfiere una alícuota de 1.00-mL a un matraz aforado de 10 mL y se diluye a volumen con metanol. Cuando se analiza por HPLC, se encuentra que la señal para la cafeína es 21 469. Reporte los miligramos de cafeína en la tableta analgésica.
24. Kagel y Farwell reportan un método HPLC de fase inversa para determinar la concentración de ácido acetilsalicílico (ASA) y cafeína (CAF) en tabletas analgésicas usando ácido salicílico (SA) como patrón interno [Kagel, R. A.; Farwell, S. O. J. Chem. Educ. 1983, 60, 163—166]. Se preparó una serie de estándares agregando cantidades conocidas de ácido acetilsalicílico y cafeína a matraces Erlenmeyer de 250 ml y agregando 100 mL de metanol. A cada una se le añadió una alícuota de 10.00 ml de una solución estándar de ácido salicílico. Se obtuvieron los siguientes resultados para un conjunto típico de soluciones estándar.
| miligramos de | cocientes de altura pico para | |||
| estándar | ASA | CAF | ASA/SA | CAF/SA |
| 1 | 200.0 | 20.0 | 20.5 | 10.6 |
| 2 | 250.0 | 40.0 | 25.1 | 23.0 |
| 3 | 300.0 | 60.0 | 30.9 | 36.8 |
Una muestra de un comprimido analgésico se colocó en un matraz Erlenmeyer de 250 ml y se disolvió en 100 mL de metanol. Después de agregar una porción de 10.00 ml del patrón interno, la solución se filtró. El análisis de la muestra dio una relación de altura máxima de 23.2 para ASA y de 17.9 para CAF.
a) Determinar los miligramos de ASA y CAF en la tableta.
b) ¿Por qué es necesario filtrar la muestra?
(c) Las indicaciones indican que se emplean aproximadamente 100 mL de metanol para disolver los patrones y muestras. ¿Por qué no es necesario medir este volumen con mayor precisión?
d) En presencia de humedad, el ASA se descompone en SA y ácido acético. ¿Qué complicación podría presentar esto para este análisis? ¿Cómo podrías evaluar si esto es un problema?
25. Bohman y colegas describieron un método de HPLC de fase inversa para el análisis cuantitativo de vitamina A en alimentos utilizando el método de adiciones estándar Bohman, O.; Engdahl, K. A.; Johnsson, H. J. Chem. Educ. 1982, 59, 251—252]. En un ejemplo típico, se coloca una muestra de 10.067-g de cereal en un matraz Erlenmeyer de 250 ml junto con 1 g de ascorbato de sodio, 40 ml de etanol y 10 ml de KOH al 50% p/v. Después de calentar a reflujo durante 30 min, se agregan 60 mL de etanol y la solución se enfría a temperatura ambiente. La vitamina A se extrae usando tres porciones de 100 mL de hexano. Las porciones combinadas de hexano se evaporan y el residuo que contiene vitamina A se transfiere a un matraz aforado de 5 mL y se diluye a volumen con metanol. Se prepara una adición estándar de manera similar usando una muestra de 10.093-g del cereal y la adición de 0.0200 mg de vitamina A. Inyectar la muestra y la adición estándar en la HPLC da áreas de pico de, respectivamente,\(6.77 \times10^3\) y\(1.32 \times 10^4\). Reporte el contenido de vitamina A de la muestra en miligramos/100 g de cereal.
26. Ohta y Tanaka reportaron un método cromatográfico de intercambio iónico para el análisis simultáneo de varios aniones inorgánicos y los cationes Mg 2 + y Ca 2 + en agua [Ohta, K.; Tanaka, K. Anal. Chim. Acta 1998, 373, 189—195]. La fase móvil incluye el ligando 1,2,4-bencenetricarboxilato, que absorbe fuertemente a 270 nm. La detección indirecta de los analitos es posible porque su absorbancia disminuye cuando se compleja con un anión.
a) El procedimiento también contempla la adición del ligando EDTA a la fase móvil. ¿Qué papel juega el EDTA en este análisis?
(b) Una solución estándar de NaHCO 3 1.0 mM, NaNo 2 0.20 mM, MgSO 4 0.20 mM, CaCl 2 0.10 mM y Ca 0.10 mM (NO 3) 2 da las siguientes áreas de pico (unidades arbitrarias).
| ion | \(\text{HCO}_3^-\) | Cl — | \(\text{NO}_2^-\) | \(\text{NO}_3^-\) |
| área de pico | 373.5 | 322.5 | 264.8 | 262.7 |
| ion | Ca 2 + | Mg 2+ | \(\text{SO}_4^{2-}\) | |
| área de pico | 458.9 | 352.0 | 341.3 |
El análisis de una muestra de agua de río (pH de 7.49) da los siguientes resultados.
| ion | \(\text{HCO}_3^-\) | Cl — | \(\text{NO}_2^-\) | \(\text{NO}_3^-\) |
| área de pico | 310.0 | 403.1 | 3.97 | 157.6 |
| ion | Ca 2 + | Mg 2+ | \(\text{SO}_4^{2-}\) | |
| área de pico | 734.3 | 193.6 | 324.3 |
Determinar la concentración de cada ion en la muestra.
(c) La detección de\(\text{HCO}_3^-\) realmente da la concentración total de carbonato en solución ([\(\text{CO}_3^{2-}\)] + [\(\text{HCO}_3^-\)] + [H 2 CO 3]). Dado que el pH del agua es de 7.49, ¿cuál es la concentración real de\(\text{HCO}_3^-\)?
(d) Un análisis independiente da las siguientes concentraciones adicionales para iones en la muestra: [Na +] = 0.60 mM; [\(\text{NH}_4^+\)] = 0.014 mM; y [K +] = 0.046 mM. El balance iónico de una solución se define como la relación entre la carga catiónica total y la carga aniónica total. Determinar el saldo de carga para esta muestra de agua y comentar si el resultado es razonable.
27. Las concentraciones de Cl —\(\text{NO}_2^-\), y\(\text{SO}_4^{2-}\) se determinan por cromatografía iónica. Una muestra estándar de 50-μL de 10.0 ppm Cl —, 2.00 ppm\(\text{NO}_2^-\) y 5.00 ppm\(\text{SO}_4^{2-}\) dio señales (en unidades arbitrarias) de 59.3, 16.1 y 6.08 respectivamente. Una muestra de efluente de una planta de tratamiento de aguas residuales se diluye diez veces y una porción de 50 μL da señales de 44.2 para Cl —, 2.73 para\(\text{NO}_2^-\) y 5.04 para\(\text{SO}_4^{2-}\). Reportan las partes por millón por cada anión en la muestra de efluente.
28. Se analizó una serie de patrones de polivinilpiridina de diferente peso molecular mediante cromatografía de exclusión por tamaño, dando los siguientes resultados.
| peso de la fórmula | volumen de retención (mL) |
|---|---|
| 600000 | 6.42 |
| 100000 | 7.98 |
| 30000 | 9.30 |
| 3000 | 10.94 |
Cuando se analiza una preparación de polivinilpiridina de peso de fórmula desconocido, el volumen de retención es de 8.45 mL. Reporte el peso promedio de la fórmula para la preparación.
29. Los refrescos dietéticos contienen cantidades apreciables de aspartamo, ácido benzoico y cafeína. Cuál es el orden esperado de elución para estos compuestos en una separación por electroforesis de zona capilar usando un tampón de pH 9.4 dado que el aspartamo tiene valores de p K a de 2.964 y 7.37, el ácido benzoico tiene un p K a de 4.2, y el p K a para la cafeína es menor que 0. La Figura 12.8.3 proporciona las estructuras de estos compuestos.
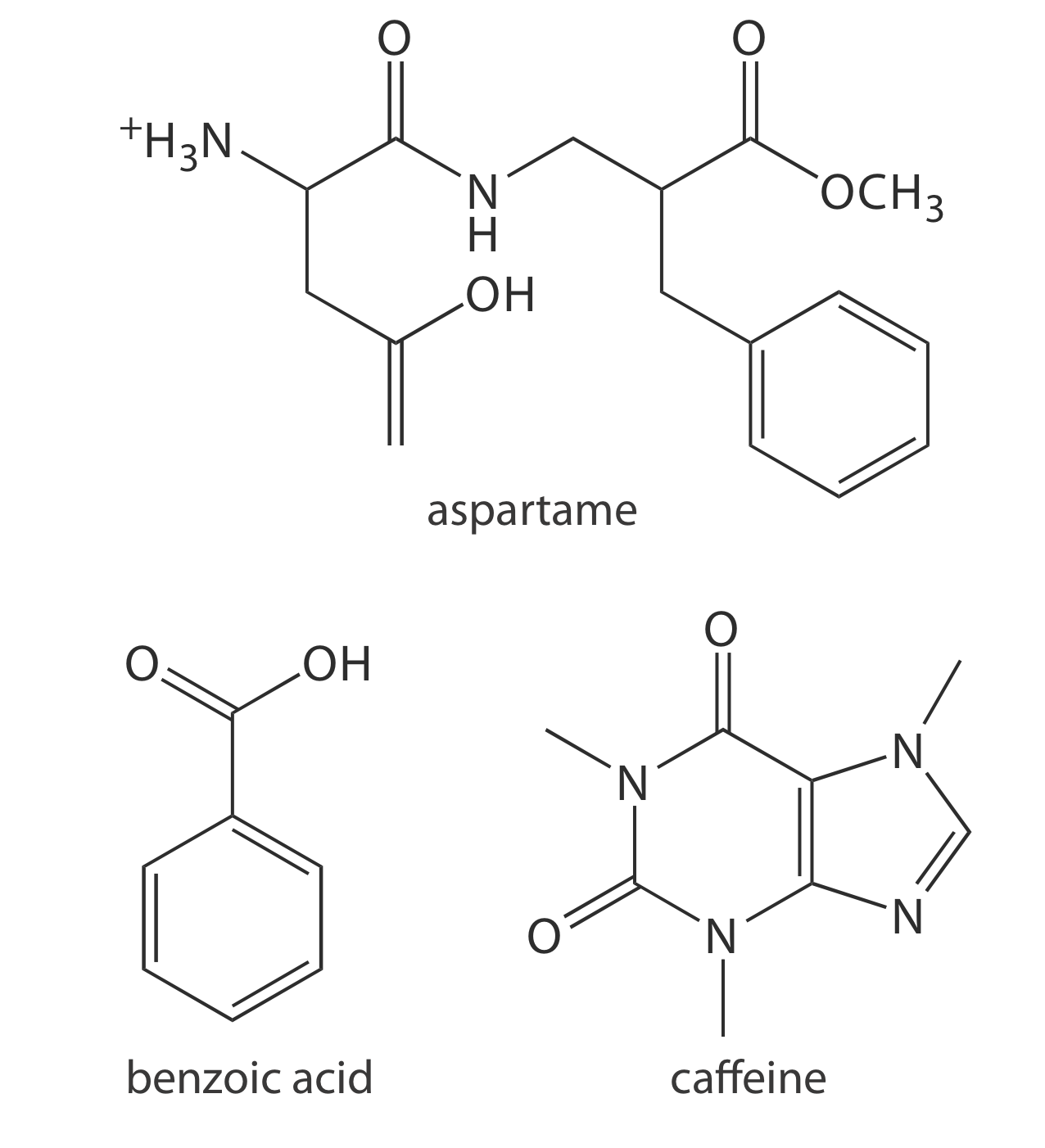 Figura 12.8.3
. Estructuras para los compuestos en el Problema 29.
Figura 12.8.3
. Estructuras para los compuestos en el Problema 29.30. Janusa y compañeros de trabajo describen la determinación de cloruro por CZE [Janusa, M. A.; Andermann, L. J.; Kliebert, N. M.; Nannie, M. H. J. Chem. Educ. 1998, 75, 1463—1465]. El análisis de una serie de estándares externos da la siguiente curva de calibración.
\[\text { area }=-883+5590 \times \mathrm{ppm} \text{ Cl}^{-} \nonumber\]
Una muestra estándar de 57.22% w/w Cl — se analiza colocando porciones de 0.1011-g en matraces volumétricos separados de 100 mL y diluyendo a volumen. Se preparan tres incógnitas pipeteando 0.250 mL, 0.500 mL, un 0.750 mL del bulto desconocido en matraces volumétricos separados de 50 mL y diluyendo a volumen. El análisis de las tres incógnitas da áreas de 15 310, 31 546 y 47 582, respectivamente. Evaluar la exactitud de este análisis.
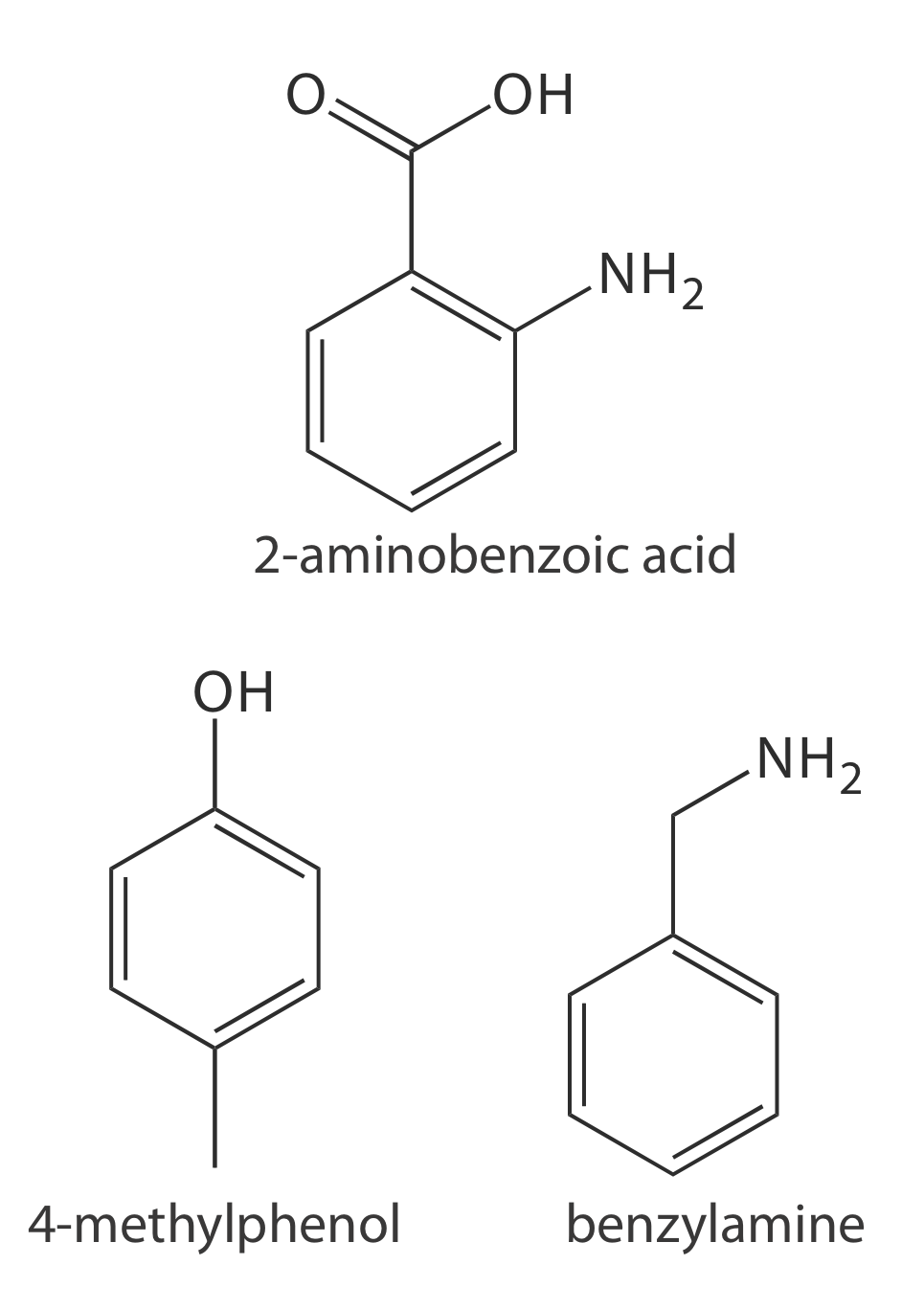
31. El análisis de agua\(\text{NO}_3^-\) en acuario se realiza por CZE utilizando\(\text{IO}_4^-\) como estándar interno. Una solución estándar de 15.0 ppm\(\text{NO}_3^-\) y 10.0 ppm\(\text{IO}_4^-\) da alturas de pico (unidades arbitrarias) de 95.0 y 100.1, respectivamente. Una muestra de agua de un acuario se diluye 1:100 y se agrega suficiente estándar interno para hacer su concentración 10.0 ppm en\(\text{IO}_4^-\). El análisis da señales de 29.2 y 105.8 para\(\text{NO}_3^-\) y\(\text{IO}_4^-\), respectivamente. Reportan las ppm\(\text{NO}_3^-\) en la muestra de agua de acuario.
32. Sugerir condiciones para separar una mezcla de ácido 2-aminbenzoico (p K a1 = 2.08, p K a2 = 4.96), bencilamina (p K a = 9.35) y 4-metilfenol (p K a2 = 10.26) por electroforesis de zona capilar. La Figura\(PageIndex{4}\) proporciona las estructuras de estos compuestos.
33. McKillop y asociados examinaron la separación electroforética de algunas alquilpiridinas por CZE [McKillop, A. G.; Smith, R. M.; Rowe, R. C.; Wren, S. A. C. Anal. Chem. 1999, 71, 497—503]. Las separaciones se realizaron utilizando capilares de 50-μm o 75-μm de diámetro interno, con una longitud total de 57 cm y una longitud de 50 cm desde el punto de inyección hasta el detector. El tampón de ejecución fue un tampón fosfato de litio pH 2.5. Las separaciones se lograron usando un voltaje aplicado de 15 kV. La movilidad electroosmótica, μ eof, medida con un marcador neutro, se encontró que era\(6.398 \times 10^{-5}\) cm 2 V —1 s —1. El coeficiente de difusión para las alquilpiridinas es\(1.0 \times 10^{-5}\) cm 2 s —1.
(a) Calcular la movilidad electroforética para 2-etilpiridina dado que su tiempo de elución es de 8.20 min.
b) ¿Cuántas placas teóricas hay para la 2-etilpiridina?
(c) Las movilidades electroforéticas para 3-etilpiridina y 4-etilpiridina son\(3.366 \times 10^{-4}\) cm 2 V —1 s —1 y\(3.397 \times 10^{-4} \text{ cm}^2 \text{ V}^{-1} \text{ s}^{-1}\), respectivamente. ¿Cuál es la resolución esperada entre estas dos alquilpiridinas?
d) Explicar las tendencias en la movilidad electroforética que se muestran en la siguiente tabla.
| alquilpiridina | \(\mu_{ep}\)(cm 2 V —1 s —1) |
| 2-metilpiridina | \(3.581 \times 10^{-4}\) |
| 2-etilpiridina | \(3.222 \times 10^{-4}\) |
| 2-propilpiridina | \(2.923 \times 10^{-4}\) |
| 2-pentilpiridina | \(2.534 \times 10^{-4}\) |
| 2-hexilpiridina | \(2.391 \times 10^{-4}\) |
e) Explicar las tendencias en la movilidad electroforética que se muestran en la siguiente tabla.
| alquilpiridina | \(\mu_{ep}\)(cm 2 V —1 s —1) |
| 2-etilpiridina | \(3.222 \times 10^{-4}\) |
| 3-etilpiridina | \(3.366 \times 10^{-4}\) |
| 4-etilpiridina | \(3.397 \times 10^{-4}\) |
f) La p K a para piridina es de 5.229. A un pH de 2.5 la movilidad electroforética de la piridina es\(4.176 \times 10^{-4}\) cm 2 V —1 s —1. ¿Cuál es la movilidad electroforética esperada si el pH del tampón de ejecución es 7.5?