
4.5: Halogenación de alcanos. Energías y Tasas de Reacciones
- Page ID
- 72769

Las economías de las naciones altamente industrializadas del mundo se basan en gran parte en la energía y los químicos producidos a partir del petróleo. Si bien los intermedios más importantes y versátiles para la conversión del petróleo en productos químicos son compuestos con dobles o triples enlaces, también es posible preparar muchas sustancias valiosas mediante reacciones de sustitución de alcanos. En tales sustituciones, se elimina un hidrógeno de una cadena carbonada y otro átomo o grupo de átomos se une en su lugar.
Un ejemplo sencillo de una reacción de sustitución es la formación de clorometano y cloro:
\[ \ce{CH_4 + Cl_2 \rightarrow CH_3Cl + HCl}\]
La ecuación para la reacción es simple, los ingredientes son baratos y el producto es útil. No obstante, si queremos decidir de antemano si tal reacción es realmente factible, tenemos que saber más. Particularmente, tenemos que saber si la reacción procede en la dirección en la que está escrita y, de ser así, si se pueden encontrar condiciones bajo las cuales procede a un ritmo conveniente. Obviamente, si se mezclara metano y cloro y descubriera que, a lo sumo, solo se producía la\(1 \%\) conversión al producto deseado y que la\(1 \%\) conversión se pudiera lograr sólo después de un día más o menos de calentamiento fuerte, esta reacción sería a la vez demasiado desfavorable y demasiado lenta para un proceso industrial.
Una forma de visualizar los problemas involucrados es con diagramas de energía, que muestran la energía en términos de alguna coordenada de reacción arbitraria que es una medida de progreso entre los estados inicial y final (Figura 4-4). Los diagramas como la Figura 4-4 pueden no resultarle familiares, y una analogía mecánica puede ser útil para proporcionar una mejor comprensión de las ideas muy importantes involucradas. Considera una caja de dos niveles que contenga varias pelotas de tenis. Un análogo a una reacción energéticamente favorable sería tener todas las bolas
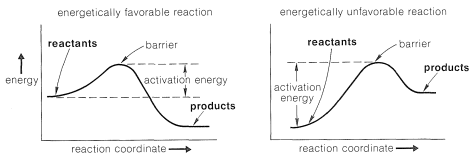
en el nivel superior donde cualquier perturbación provocaría que bajaran al nivel inferior bajo la influencia de la gravedad, perdiendo así energía.
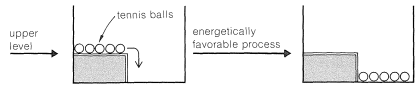
Si se modifica el nivel superior y se agrega una cerca baja para mantener las bolas en su lugar, será tan energéticamente favorable como cuando la barda no está ahí para que las bolas estén en el nivel inferior. La diferencia es que el proceso no ocurrirá espontáneamente sin alguna perturbación mayor. Podemos decir que existe una barrera energética ante la ocurrencia del proceso favorable.
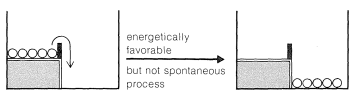
Ahora bien, si agitamos la caja lo suficientemente fuerte, las bolas en el nivel superior pueden adquirir suficiente energía para rebotar sobre la barrera y caer al nivel inferior. Entonces se puede decir que las bolas adquieren suficiente energía de activación para superar la barrera. A nivel molecular, la energía de activación debe ser adquirida ya sea por colisiones entre moléculas como resultado de sus movimientos térmicos, o de alguna agencia externa, para permitir que los reactivos superen la barrera y se transformen en productos. En breve discutiremos esto más, pero primero queremos ilustrar otro concepto importante con nuestra analogía mecánica, la del equilibrio y el equilibrio.
Con un suave temblor de nuestra caja de dos niveles, se espera que todas las bolas del nivel superior terminen en el nivel inferior. No habrá suficiente activación para que pasen del nivel inferior al superior. En esta circunstancia, podemos decir que las bolas no están equilibradas entre los niveles inferior y superior. No obstante, si agitamos la caja vigorosa y continuamente, no importa si empezamos con todas las bolas en el nivel inferior o superior, se establecerá un equilibrio con, en promedio, la mayoría de las bolas en el nivel inferior energéticamente más favorable, pero algunas en el nivel superior también.
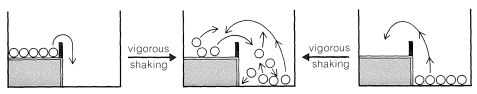
Para mantener una fracción promedio constante de las bolas en cada nivel con agitación vigorosa y continua, la velocidad a la que las bolas van del nivel superior al nivel inferior debe ser igual a la velocidad que van en sentido contrario. Ahora las bolas se equilibrarán entre los dos niveles. En equilibrio, la fracción de las bolas en cada uno de los dos niveles es totalmente independiente de la altura de la barrera, siempre y cuando la activación (agitación) sea suficiente para permitir que las bolas vayan en ambos sentidos.
Los diagramas de la Figura 4-4 deben interpretarse de la misma manera general. Si la agitación térmica de las moléculas es suficiente, entonces se puede esperar que se establezca un equilibrio entre los reactivos y los productos, ya sea que la reacción global sea energéticamente favorable (lado izquierdo de la Figura 4-4) o energéticamente desfavorable (lado derecho de la Figura 4-4). Pero al igual que con nuestra analogía, cuando se establece el equilibrio esperamos que la mayor parte de las moléculas esté en el estado energético más favorable.
¿Qué sucede cuando el metano se mezcla con cloro? No se produce ninguna reacción medible cuando los gases se mezclan y se mantienen en la oscuridad a temperatura ambiente. Claramente, o bien la reacción es energéticamente desfavorable o la barrera energética es alta. La respuesta en cuanto a cuál se aclara cuando la mezcla se calienta a temperaturas superiores\(300^\text{o}\) o cuando se expone a una fuerte luz violeta o ultravioleta, por lo que se produce una reacción rápida o incluso explosiva. Por lo tanto, la reacción es energéticamente favorable, pero la energía de activación es mayor que la que se puede lograr por agitación térmica sola a temperatura ambiente. Por lo tanto, el calor o la luz deben iniciar una vía para que los reactivos se conviertan en productos que tengan una barrera baja o energía de activación.
¿Podríamos haber predicho los resultados de este experimento antes de tiempo? Primero, debemos reconocer que aquí realmente hay varias preguntas. ¿Podríamos haber decidido si la reacción fue energéticamente favorable? ¿Que la reacción oscura sería lenta a temperatura ambiente? ¿Esa luz haría que la reacción fuera rápida? Consideramos estas y algunas preguntas relacionadas en detalle porque son preguntas importantes y las respuestas a las mismas son relevantes de una manera u otra para el estudio de todas las reacciones en química orgánica.
La cuestión de la constante de equilibrio
Presumiblemente, el metano podría reaccionar con el cloro para dar clorometano y cloruro de hidrógeno, o el clorometano podría reaccionar con cloruro de hidrógeno para dar metano y cloro. Si se encontraran condiciones para las cuales ambas reacciones procedieran a una velocidad finita, finalmente se establecería el equilibrio cuando las velocidades de las reacciones en cada dirección fueran iguales:
\[ \ce{CH_4 + Cl_2 \rightleftharpoons CH_3Cl + HCl}\]
En equilibrio, la relación entre las cantidades de reactivos y productos viene dada por la expresión constante de equilibrio
\[K_{eq} = \dfrac{[CH_3Cl][HCl]}{[CH_4][Cl_2]} \label{4-1}\]
en la que\(K_\text{eq}\) se encuentra la constante de equilibrio.
Las cantidades dentro de los paréntesis de la Ecuación\(\ref{4-1}\) denotan concentraciones para reactivos líquidos o presiones parciales para sustancias gaseosas. Si la constante de equilibrio\(K_\text{eq}\) es mayor que\(1\), entonces al mezclar volúmenes iguales de cada una de las sustancias participantes (todas son gases arriba\(-24^\text{o}\)), la reacción a la derecha será inicialmente más rápida que la reacción a la izquierda, hasta que se establezca el equilibrio; en este punto habrá más clorometano y cloruro de hidrógeno presentes que metano y cloro. Sin embargo, si la constante de equilibrio fuera menor que\(1\), la reacción inicialmente procedería más rápido a la izquierda y, en equilibrio, habría más metano y cloro presentes que el clorometano y el cloruro de hidrógeno. \(^4\)Para la cloración de metano, sabemos por experimento que la reacción va a la derecha y eso\(K_\text{eq}\) es mucho mayor que la unidad. Naturalmente, sería útil en la planeación de otras preparaciones orgánicas para poder estimar con\(K_\text{eq}\) anticipación.
Es una experiencia común asociar reacciones químicas con constantes de equilibrio mayores a una con la evolución del calor, es decir, con\(\Delta H^\text{0}\) valores negativos. Hay, de hecho, muchos ejemplos llamativos. La formación de clorometano y cloruro de hidrógeno a partir de metano y cloro tiene un\(K_\text{eq}\)\(\Delta H^\text{0}\) de\(10^{18}\) y de\(-24 \: \text{kcal}\) por mol de\(CH_3Cl\) formado a\(25^\text{o}\). La combustión de hidrógeno con oxígeno para dar agua tiene un\(K_\text{eq}\) de\(10^{40}\) y\(\Delta H^\text{0} = -57 \: \text{kcal}\) por mol de agua formada en\(25^\text{o}\). Sin embargo, esta correlación entre\(K_\text{eq}\) y no\(\Delta H^\text{0}\) es universal ni rigurosa. Se conocen reacciones que absorben calor (son endotérmicas) y aún así tienen\(K_\text{eq} > 1\). Otras reacciones tienen\(\Delta H^\text{0}\) valores grandes y constantes de equilibrio mucho menores que\(1\).
El problema es que el cambio energético con el que se correlaciona no\(K_\text{eq}\) es\(\Delta H^\text{0}\) sino\(\Delta G^\text{0}\) (el llamado cambio de "energía estándar de Gibbs “)\(^5\), y si sabemos\(\Delta G^\text{0}\), podemos calcular\(K_\text{eq}\) por la ecuación
\[ \Delta G^o =-2.303 RT \log_{10} K_{eq} \label{4-2}\]
en el que\(R\) es la constante del gas y\(T\) es la temperatura absoluta en grados Kelvin. Para nuestros cálculos, vamos a utilizar\(R\) como\(1.987 \: \text{cal} \: \text{deg}^{-1} \: \text{mol}^{-1}\) y no debes olvidar convertir\(\Delta G^\text{0}\) a\(\text{cal}\).
Las tablas de\(\Delta G^\text{0}\) valores para la formación de compuestos particulares (a diversas temperaturas y estados) a partir de los elementos están disponibles en los manuales y la literatura. Con estos, podemos calcular las constantes de equilibrio con bastante precisión. Por ejemplo, los manuales dan los siguientes datos, que son útiles para la cloración del metano:
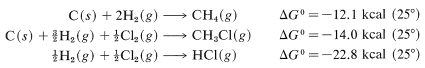
Combinando estos con la debida consideración por la señal da
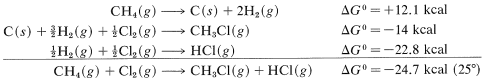
y\(\text{log} \: K_\text{eq} = -\left( -24.7 \times 1000 \right)/ \left(2.303 \times 1.987 \times 298.2 \right)\), entonces\(K_\text{eq} = 1.3 \times 10^{18}\). Desafortunadamente,\(\Delta G^\text{0}\) los valores insuficientes para las reacciones de formación están disponibles para hacer de este un método ampliamente aplicable para calcular\(K_\text{eq}\) valores.
La situación no es totalmente desesperada, porque existe una relación entre\(\Delta G^\text{0}\) y\(\Delta H^\text{0}\) que también implica\(T\) y otra cantidad,\(\Delta S^\text{0}\), el cambio de entropía estándar del proceso:
\[ \Delta G^o = \Delta H^o -T \Delta S^o \label{4-3}\]
Esta ecuación muestra que\(\Delta G^\text{0}\) y\(\Delta H^\text{0}\) son iguales cuando\(\Delta S^\text{0}\) es cero. Por lo tanto, el signo y la magnitud de\(T \Delta S^\text{0}\) determinar qué tan bien\(K_\text{eq}\) se correlaciona con\(\Delta H^\text{0}\). Ahora, tenemos que prestar atención a si podemos estimar\(T \Delta S^\text{0}\) los valores lo suficientemente bien como para decidir si el\(\Delta H^\text{0}\) de una reacción dada (calculada a partir de energías de enlace u otra información) dará una buena o mala medida de\(\Delta G^\text{0}\).
Entropía y Trastorno Molecular
Para decidir si necesitamos preocuparnos\(\Delta S^\text{0}\) con respecto a alguna reacción en particular, tenemos que tener alguna idea de qué significado físico tiene la entropía. Ser muy detallado sobre este tema está más allá del alcance de este libro, pero debes tratar de entender la base física de la entropía, porque si lo haces, entonces podrás predecir al menos cualitativamente si\(\Delta H^\text{0}\) será aproximadamente la misma o muy diferente de\(\Delta G^\text{0}\). Esencialmente, la entropía de un sistema químico es una medida de su desorden molecular o aleatoriedad. Al igual que otras cosas, cuanto más aleatorio es el sistema, más favorable es el sistema.
Diferentes tipos de moléculas tienen diferentes grados de libertad de traducción, vibración y rotación y, por lo tanto, diferentes grados promedio de trastorno molecular o aleatoriedad. Ahora bien, si para una reacción química el grado de trastorno molecular es diferente para los productos que para los reactivos, habrá un cambio en la entropía y\(\Delta S^\text{0} \neq 0\).
Un ejemplo espectacular del efecto del trastorno molecular en contribuir a la diferencia entre\(\Delta H^\text{0}\) y\(\Delta G^\text{0}\) es proporcionado por la formación de nonano líquido,\(C_9H_{20}\), a partir de carbono sólido y gas hidrógeno en\(25^\text{o}\):
\[\ce{9C(s) + 10H_2(g) \rightarrow C_910_{20}(l)}\]
con\(\Delta H^o = -54.7 \, kcal\) y\(\Delta S^o = 5.0 \, kcal\).
Ecuaciones\(\ref{4-2}\) y se\(\ref{4-3}\) pueden reorganizar para calcular\(\Delta S^\text{0}\) y\(K_\text{eq}\) desde\(\Delta H^\text{0}\) y\(\Delta G^\text{0}\):
y
\[K_{eq} = 10^{-\Delta G^o/2.303 \,RT} = 10^{-5.900/(2.303 \times 1.987 \times 298.2)} = 4.7 \times 10^{-5}\]
Estos\(\Delta H^\text{0}\),\(\Delta S^\text{0}\), y\(K_\text{eq}\) los valores se pueden comparar con aquellos para\(H_2 + \frac{1}{2} O_2 \longrightarrow H_2O\), para los cuales\(\Delta H^\text{0}\) es\(-57 \: \text{kcal}\),\(\Delta S^\text{0}\) es\(8.6 \: \text{e.u.}\), y\(K_\text{eq}\) es\(10^{40}\). Obviamente, hay algo desfavorable en el cambio de entropía de carbono e hidrógeno a nonano. Lo importante es que hay una gran diferencia en las restricciones sobre los átomos a cada lado de la ecuación. En particular, las moléculas de hidrógeno en estado gaseoso tienen una gran libertad de traducción y un alto grado de desorden, la mayor parte del cual se pierde cuando los átomos de hidrógeno se unen a una cadena de carbonos. Esto lo convierte en un gran negativo\(\Delta S^\text{0}\), lo que corresponde a una disminución de\(K_\text{eq}\). Las diferencias en las restricciones de los carbonos son menos importantes. El carbono sólido tiene una estructura ordenada y rígida con poca libertad de movimiento de los átomos de carbono individuales. Estos carbonos están menos restringidos en el nonano, y esto tendería a hacer\(\Delta S^\text{0}\) más positivos y\(\Delta G^\text{0}\) más negativos, lo que corresponde a un aumento de\(K_\text{eq}\) (Ecuaciones\(\ref{4-2}\) y\(\ref{4-3}\)). Sin embargo, este es un pequeño efecto en\(\Delta S^\text{0}\) comparación con la enorme diferencia en el grado de desorden del hidrógeno entre el gas hidrógeno y el hidrógeno unido al carbono en el nonano.
Los efectos negativos de entropía generalmente se observan en reacciones de cierre de anillo como la formación de ciclohexano a partir del 1-hexeno, que ocurren con pérdida sustancial de libertad rotacional (trastorno) sobre los\(C-C\) enlaces:
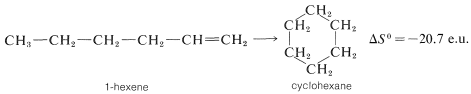
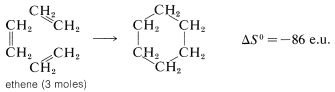
Hay una pérdida aún mayor en la entropía al formar ciclohexano a partir del eteno porque se pierde sustancialmente más libertad en la orientación de tres moléculas de eteno para formar un anillo:
Para reacciones simples, con el mismo número de moléculas en cada lado de la ecuación, sin formación de anillos u otros cambios inusuales en las restricciones entre los productos y reactivos,\(\Delta S^\text{0}\) generalmente es relativamente pequeño. En general, para este tipo de procesos, sabemos por experiencia que\(K_\text{eq}\) por lo general es mayor que 1 si\(\Delta H^\text{0}\) es más negativo que\(-15 \: \text{kcal}\) y por lo general es menor que 1 para\(\Delta H^\text{0}\) más positivo que\(+15 \: \text{kcal}\). Podemos usar esto como una “regla general” para predecir si\(K_\text{eq}\) debe ser mayor o menor que la unidad para reacciones en fase vapor que involucran moléculas simples. Alguna idea del grado de éxito que se espera de esta regla puede inferirse de los ejemplos del Cuadro 4-5, que también contiene una comparación adicional de algunos\(\Delta H^\text{0}\) valores experimentales con los calculados a partir de energías de enlace.
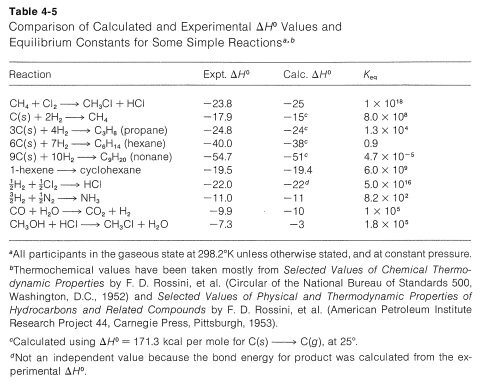
Supongamos que\(\Delta G^\text{0}\) es positivo, ¿qué esperanza tenemos de obtener una conversión útil a un producto deseado? No hay una respuesta simple, directa y general a esta pregunta. Cuando la reacción es reversible el procedimiento clásico de eliminar uno o más de los productos para evitar que se establezca el equilibrio tiene muchas aplicaciones en la química orgánica, como se verá más adelante. Cuando este enfoque es inaplicable, es necesario un cambio en los reactivos. Así, el yodo no da una conversión útil con 2,2-dimetilpropano,\(1\), para dar 1-yodo-2,2-dimetilpropano,\(2\), debido a que la posición de equilibrio está demasiado lejos a la izquierda (\(K_\text{eq} \cong 10^{-5}\)):
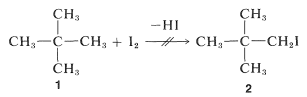
Se requieren rutas alternativas con\(\Delta G^\text{0}\) valores favorables. El desarrollo de formas de hacer indirectamente, por procesos eficientes, lo que no se puede hacer directamente es una de las actividades más interesantes y desafiantes de los químicos orgánicos.
¿Por qué el metano y el cloro no reaccionan en la oscuridad\(25^\text{o}\)?
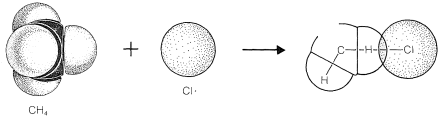
Para llegar a comprender por qué el metano y el cloro no reaccionan en la oscuridad, debemos considerar los detalles de cómo ocurre la reacción, es decir, el mecanismo de reacción. El mecanismo más simple sería que una molécula de cloro colisionara con una molécula de metano de tal manera que se formaran clorometano y cloruro de hidrógeno directamente como resultado de una ruptura concertada de los\(C-H\) enlaces\(Cl-Cl\) y y la fabricación de los\(H-Cl\) enlaces\(C-Cl\) y ( ver Figura 4-5). La falta de reacción indica que debe haber una barrera energética demasiado alta para que este mecanismo funcione. ¿Por qué debería ser así?
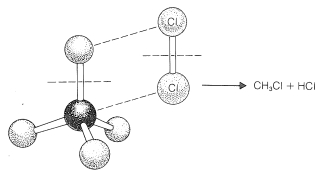
En primer lugar, este mecanismo implica una colisión de “cuatro centros” orientada con mucha precisión entre cloro y metano que tendría una baja probabilidad de ocurrencia (es decir, una gran disminución de la entropía porque una orientación precisa significa un alto orden molecular). Segundo, requiere empujar una molécula de cloro suficientemente profundamente en una molécula de metano para que uno de los átomos de cloro se acerque lo suficiente al carbono para formar un enlace y producir clorometano.
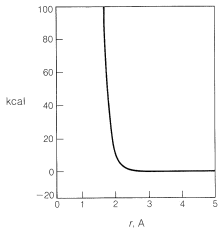
Generalmente, para llevar los átomos no unidos a distancias cercanas (\(1.2 \: \text{A}\)a\(1.8 \: \text{A}\)) de enlace requiere un gran gasto de energía, como se puede ver en la Figura 4-6. Las fuerzas repulsivas interatómicas aumentan rápidamente a distancias cortas, y empujar una molécula de cloro a una molécula de metano para alcanzar distancias similares a la distancia de enlace\(1.77\) -\(\text{A}\) carbono-cloro en el clorometano requeriría una cantidad considerable de compresión (ver Figura 4-7). Información valiosa
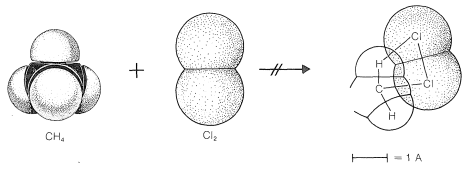
acerca de las repulsiones interatómicas se pueden obtener con modelos de relleno de espacio del tipo CPK (Sección 2-2), que tienen radios escalados para corresponder a radios de interferencia atómica reales, es decir, la distancia interatómica en el punto donde las curvas del tipo de Figura 4-6 comienzan a elevarse pronunciadamente. Con tales modelos, el grado de compresión atómica requerido para llevar los átomos no unidos a una distancia cercana a la unión es más evidente que con los modelos de bola y varilla. Cabe señalar que las reacciones de cuatro centros del tipo postulado en la Figura 4-5 se encuentran solo en raras ocasiones.
Si se descarta el mecanismo concertado de cuatro centros para la formación de clorometano y cloruro de hidrógeno a partir del cloro y el metano, todas las posibilidades restantes son mecanismos de reacción paso a paso. Una reacción lenta paso a paso es dinámicamente análoga al flujo de arena a través de una sucesión de embudos con diferentes diámetros de tallo. El embudo con el tallo más pequeño será el cuello de botella más importante y, si su diámetro de tallo es mucho menor que los demás, solo determinará el caudal. Generalmente, una reacción química de múltiples etapas tendrá una etapa de determinación de velocidad lenta (análoga al embudo con el tallo pequeño) y otras etapas relativamente rápidas, que pueden ocurrir antes o después de la etapa lenta.
Un posible conjunto de etapas para la cloración del metano sigue:

Las reacciones (1) y (2) implican la disociación del cloro en átomos de cloro y la ruptura de un\(C-H\) enlace de metano para dar un radical metilo y un átomo de hidrógeno. El radical metilo, al igual que los átomos de cloro e hidrógeno, tiene una elección no involucrada en el enlace. Los átomos y radicales suelen ser altamente reactivos, por lo que la formación de clorometano y cloruro de hidrógeno debe proceder fácilmente por las Reacciones (3) y (4). El quid entonces será si las etapas (1) y (2) son razonables bajo las condiciones de reacción.
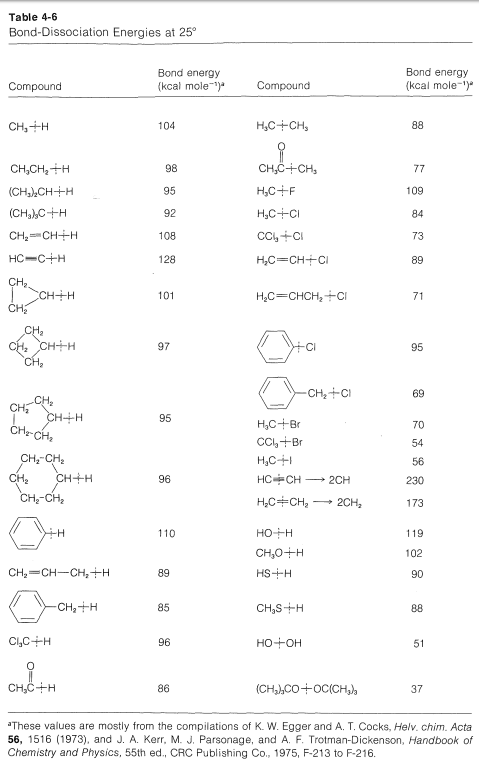
En ausencia de algún estímulo externo, solo las colisiones debidas a los movimientos térmicos habituales de las moléculas pueden proporcionar la energía necesaria para romper los enlaces. A temperaturas inferiores\(100^\text{o}\), es muy raro que la agitación térmica por sí sola pueda suministrar suficiente energía para romper cualquier número significativo de enlaces más fuertes que\(30\) a\(35 \: \text{kcal mol}^{-1}\).
La energía de\(Cl-Cl\) unión de la Tabla 4-3 es\(58.1 \: \text{kcal}\), que es demasiado grande para permitir la ruptura del enlace por agitación\(25^\text{o}\) térmica de acuerdo con la Reacción (1). Para la Reacción (2) no es recomendable utilizar la energía de\(98.7 \: \text{kcal} \: C-H\) enlace del Cuadro 4-3 porque esta es una cuarta parte de la energía requerida para romper los cuatro\(C-H\) enlaces (Sección 4-3). Energías de disociación de enlace más específicas se dan en la Tabla 4-5, y se verá que para romper un\(C-H\) enlace de metano se requiere\(104 \: \text{kcal}\) at\(25^\text{o}\), que de nuevo es demasiado para ser ganado por agitación térmica. Por lo tanto podemos concluir que las Reacciones (1) - (4) no pueden ser un mecanismo importante para la cloración del metano a temperatura ambiente.
Uno podría preguntarse si la disociación en iones proporcionaría mecanismos viables para la cloración del metano. Parte de la respuesta ciertamente es: No en fase vapor, como muestran los siguientes datos termoquímicos:
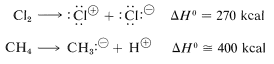
La disociación iónica simplemente no ocurre a temperaturas normalmente accesibles por colisiones entre moléculas en estado de vapor. Lo que se necesita para la formación de iones es un estímulo externo altamente energético, como el bombardeo con electrones de rápido movimiento, o un disolvente ionizante que ayude a la ionización. Ambos procesos serán discutidos más adelante. El punto aquí es que la disociación iónica no es un paso viable para la cloración de metano en fase vapor.
¿Por qué la luz induce la cloración del metano?
Primero, debemos dejar claro que la luz hace más que proporcionar energía simplemente para levantar las moléculas de metano y cloro sobre la barrera de la Figura 4-4. Esto es evidente por el hecho de que se necesita muy poca luz, mucho menos de un fotón de luz por molécula de clorometano producida. La luz podría activar metano o cloro, o ambos. Sin embargo, el metano es incoloro y el cloro es amarillo-verde. Esto indica que el cloro, no el metano, interactúa con la luz visible. Un fotón de luz casi ultravioleta, tal como es absorbido por el gas cloro, proporciona energía más que suficiente para dividir la molécula en dos átomos de cloro:
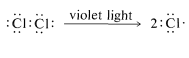
Una vez producido, un átomo de cloro puede eliminar un átomo de hidrógeno de una molécula de metano y formar un radical metilo y una molécula de cloruro de hidrógeno. Las energías de enlace disociación de\(CH_4\) (\(104 \: \text{kcal}\)) y\(HCl\) (\(103.1 \: \text{kcal}\)) sugieren que esta reacción es endotérmica por aproximadamente\(1 \: \text{kcal}\):
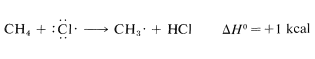
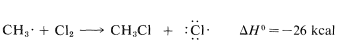
El uso de energías de enlace disociación da un cálculo\(\Delta H^\text{0}\) de\(-26 \: \text{kcal}\) para esta reacción, que sin duda es lo suficientemente grande, según nuestra regla general, para predecir que\(K_\text{eq}\) será mayor que 1. Se espera que el ataque de un radical metilo sobre el cloro molecular requiera una colisión algo más orientada que para un átomo de cloro que reacciona con el metano (la molécula de cloro probablemente debería estar en sentido extremo, no lateral, hacia el radical) pero la repulsión interatómica probablemente no debería ser muy diferente.
El resultado neto de\(CH_4 + Cl \cdot \longrightarrow CH_3 \cdot + HCl\) y\(CH_3 \cdot + Cl_2 \longrightarrow CH_3Cl + Cl \cdot\) es la formación de clorometano y cloruro de hidrógeno a partir de metano y cloro. Observe que el átomo de cloro consumido en el primer paso es reemplazado por otro en el segundo paso. Este tipo de secuencia de reacciones se denomina reacción en cadena porque, en principio, un átomo puede inducir la reacción de un número infinito de moléculas a través de la operación de una “cadena” o ciclo de reacciones. En nuestro ejemplo, los átomos de cloro formados por la acción de la luz
\(Cl_2\)puede inducir la cloración del metano mediante las etapas de propagación en cadena:
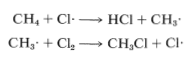
En la práctica, las reacciones en cadena están limitadas por los llamados procesos de terminación. En nuestro ejemplo, los átomos de cloro o los radicales metilo se destruyen al reaccionar entre sí, como se muestra en las siguientes ecuaciones:
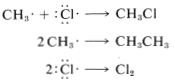
Se puede considerar que las reacciones en cadena involucran tres fases: Primero, debe ocurrir la iniciación de la cadena, que para la cloración del metano es la activación y conversión de moléculas de cloro en átomos de cloro por la luz. En segundo lugar, los pasos de propagación en cadena convierten los reactivos en productos sin consumo neto de átomos o radicales. Las reacciones de propagación ocurren en competencia con etapas de terminación de cadena, que resultan en la destrucción de átomos o radicales. Armando todo, podemos escribir:
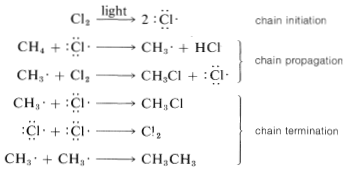
Se espera que las reacciones de terminación de cadena sean extremadamente rápidas porque los átomos y los radicales tienen electrones en conchas sin llenar que normalmente se unen. Como resultado, la formación de enlaces puede comenzar tan pronto como los átomos o radicales se acercan entre sí de cerca, sin necesidad de que otros enlaces comiencen a romperse. La evidencia es fuerte de que las reacciones formadoras de enlaces entre átomos y radicales generalmente están controladas por difusión, que casi no se requiere energía de barrera o activación, y las velocidades de combinación son simplemente las velocidades a las que ocurren los encuentros entre radicales o átomos.
Si las tasas de combinación de radicales o átomos son tan rápidas, bien podría preguntarse cómo podría competir la propagación de cadenas. Por supuesto, la competencia será posible si las reacciones de propagación en sí son rápidas, pero otra consideración importante es el hecho de que las concentraciones de átomos o radicales son muy bajas. Supongamos que la concentración de\(Cl \cdot\) es\(10^{-11} \: \text{M}\) y la\(CH_4\) concentración\(1 \: \text{M}\). La probabilidad de encuentros entre dos\(Cl \cdot\) átomos será proporcional a\(10^{-11} \times 10^{-11}\),\(CH_4\) y entre y\(Cl \cdot\) átomos será\(10^{-11} \times 1\). Así, siendo otras cosas iguales,\(CH_4 + Cl \cdot \longrightarrow CH_3 \cdot + HCl\) (propagación) se vería favorecida sobre\(2Cl \cdot \longrightarrow Cl_2\) (terminación) por un factor de\(10^{11}\). En condiciones favorables, la cadena de metano-cloración puede pasar por 100 a 10,000 ciclos antes de que se produzca la terminación por combinación radical o de átomos. En consecuencia, la eficiencia (o rendimiento cuántico) de la reacción es muy alta en términos de la cantidad de cloración que se produce en relación con la cantidad de luz absorbida.
Las tasas generales de reacciones en cadena suelen ser muy ralentizadas por sustancias que pueden combinarse con átomos o radicales y convertirlos en especies incapaces de participar en las etapas de propagación en cadena. Tales sustancias se llaman trampas de radicales, o inhibidores. El oxígeno actúa como inhibidor en la cloración del metano al combinarse rápidamente con un radical metilo para formar el radical peroximetilo comparativamente estable (menos reactivo),\(CH_3OO \cdot\). Esto efectivamente termina la cadena:
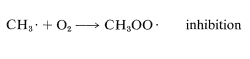
¿Podemos predecir si las reacciones serán rápidas o lentas?
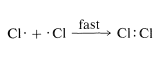
En un grado considerable, podemos predecir reactividades relativas, siempre que usemos el sentido común para limitar nuestros esfuerzos a situaciones razonables. En la sección anterior, argumentamos que las reacciones en las que se combinan átomos o radicales se pueden esperar que sean extremadamente rápidas debido a que cada entidad tiene un electrón potencialmente enlazante en una capa externa sin relleno, y unirlos para formar un enlace no requiere que se rompan otros enlaces:
La diferencia entre la energía promedio de los reactivos y la energía del estado de transición se denomina energía de activación (Figura 4-4). Esperamos que esta energía sea más pequeña (barrera inferior) si se está rompiendo un vínculo débil y se está haciendo un vínculo fuerte. El lector perceptivo notará que estamos sugiriendo un paralelo entre la velocidad de reacción y\(\Delta H^\text{0}\) porque\(\Delta H^\text{0}\) depende de la diferencia en las fortalezas de los enlaces que se rompen y forman. Sin embargo, anteriormente (Sección 4-4A), señalamos que la barrera energética para una reacción no necesita tener relación con cuán energéticamente factible es la reacción, y esto es cierto para reacciones complejas que involucran muchos pasos. Pero nuestro paralelo intuitivo entre tarifa y\(\Delta H^\text{0}\) suele funcionar bastante bien para las tasas de pasos individuales. Esto lo demuestran los datos experimentales sobre las tasas de eliminación de un átomo de hidrógeno del metano por átomos o radicales (\(X \cdot\)), tales como,,,\(F \cdot\),,,\(Cl \cdot\),\(Br \cdot\),\(HO \cdot\),,\(H_2N \cdot\),, que generalmente son paralelos a la fuerza del nuevo enlace formado:
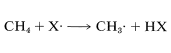
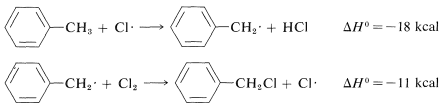
De igual manera, si observamos las energías de\(H-C\) unión-disociación de los hidrocarburos que se muestran en la Tabla 4-6,\(Cl \cdot\) inferiríamos que eliminaría un hidrógeno más rápidamente del carbono formando el\(C-H\) enlace más débil y, nuevamente, esto está muy de acuerdo con la experiencia. Por ejemplo, la cloración del metilbenceno (tolueno) a la luz solar conduce a la sustitución de un hidrógeno de metilo en lugar de un hidrógeno de anillo por la razón de que los\(C-H\) enlaces metilo son más débiles y son atacados más rápidamente que los\(C-H\) enlaces del anillo. Esto se puede ver explícitamente en\(\Delta H^\text{0}\) los valores para los pasos de propagación de cadena calculados a partir de las energías de disociación de enlace de la Tabla 4-6.
Sustitución de metilo (observada):
Sustitución del anillo (no observada):
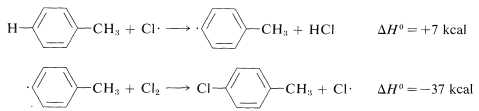
La abstracción\(\Delta H^\text{0}\) de anillo-hidrógeno es desfavorable por\(+7 \: \text{kcal}\) debido a la alta energía de\(C-H\) enlace (\(110 \: \text{kcal}\)). Por lo tanto, no se observa este paso. Es demasiado lento en comparación con la reacción más favorable en el grupo metilo a pesar de que la segunda etapa de propagación es energéticamente favorable\(-37 \: \text{kcal}\) y presumiblemente ocurriría muy rápidamente. El uso de energías de disociación de enlaces para predecir las velocidades de reacción relativas se vuelve mucho menos válido cuando tratamos de comparar diferentes tipos de reacciones. Para ilustrar, el etano podría reaccionar\(F \cdot\) para dar fluorometano o fluoruro de hidrógeno:
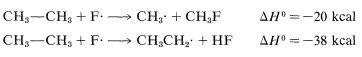
No es buena idea tratar de predecir las tasas relativas de estas dos reacciones sobre la base de sus\(\Delta H^\text{0}\) valores generales porque la naturaleza de los bonos hechos y rotos es demasiado diferente.
¿Cómo debemos hacer para formular un mecanismo de reacción?
Ante proponer un mecanismo para una reacción que implica hacer o romper en general más de dos vínculos, el principiante trata casi invariablemente de inventar un proceso en el que, con un solo paso, se rompen todos los vínculos correctos y se forman todos los vínculos correctos. Dichos mecanismos, llamados mecanismos concertados, tienen tres desventajas. En primer lugar, son casi imposibles de demostrar que son correctos. En segundo lugar, la predicción de las tasas relativas de reacciones que involucran mecanismos concertados es especialmente difícil. Tercero, los mecanismos concertados tienen cierta esterilidad ya que uno no tiene control sobre lo que sucede mientras se están llevando a cabo, excepto un control general de la tasa mediante la regulación de concentraciones, temperatura, presión, elección de solventes, etc.
Para ilustrar, supongamos que la cloración del metano parece proceder a través de un mecanismo concertado de un solo paso:
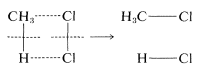
En el instante de la reacción, las moléculas reaccionantes en efecto desaparecerían en un armario oscuro y luego emergerían como moléculas de producto. No hay manera de probar experimentalmente que todos los enlaces se hicieron y formaron simultáneamente. Todo lo que se podría hacer sería utilizar las pruebas más buscadas posibles para sondear la existencia de pasos discretos. Si estas pruebas fallan, la reacción aún no se probaría concertada porque más tarde podrían desarrollarse otras, aún más pruebas de búsqueda que darían una respuesta diferente. El hecho es que una vez que aceptas que una reacción en particular es concertada, en efecto, aceptas la proposición de que seguir trabajando en su mecanismo es inútil, por importante que sientas que otros estudios serían con respecto a los factores que afectan la velocidad de reacción.
El practicante experimentado en mecanismos de reacción acepta un mecanismo concertado para una reacción que implica romper y hacer más de dos vínculos como último recurso. Primero intentará analizar la transformación global en términos de pasos discretos que son individualmente lo suficientemente simples seguramente para ser concertados y que también involucra intermedios energéticamente razonables.
Tal análisis de una reacción en términos de pasos mecanicistas discretos ofrece muchas posibilidades para estudios experimentales, especialmente en el desarrollo de procedimientos para detectar la existencia, aunque sea altamente transitoria, de los intermedios propuestos. Daremos muchos ejemplos de la fecundidad de este tipo de enfoques en discusiones posteriores.
\(^4\)Si no le son familiares los cálculos basados en constantes de equilibrio químico, le sugerimos que estudie uno de los textos de química general enumerados para lectura suplementaria al final del Capítulo 1.
\(^5\)Muchos libros y referencias utilizan\(\Delta F^\text{0}\) en lugar de\(\Delta G^\text{0}\). La diferencia entre la energía estándar de Gibbs\(\Delta G^\text{0}\) y la energía Gibbs\(\Delta G\)\(\Delta G^\text{0}\) es que se define como el valor de la energía libre cuando todos los participantes están en estados estándar. La energía libre\(\Delta G\) para una reacción\(\text{A} + \text{B} + \cdots \longrightarrow \text{X} + \text{Y} + \cdots\) es igual a\(\Delta G^\text{0} - 2.303 RT \: \text{log} \: \frac{\left[ \text{X} \right] \left[ \text{Y} \right] \cdots}{\left[ \text{A} \right] \left[ \text{B} \right] \cdots}\) donde los productos\(\left[ \text{X} \right], \left[ \text{Y} \right] \cdots\), y los reactivos\(\left[ \text{A} \right], \left[ \text{B} \right] \cdots\), no tienen que estar en estados estándar. Vamos a utilizar sólo\(\Delta G^\text{0}\) en este libro.
\(^6\)La unidad de entropía\(\text{e.u.}\) tiene las dimensiones caloría por grado o\(\text{cal deg}^{-1}\).
Colaboradores y Atribuciones
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."