
10.7: Espectroscopia de Emisión Atómica




- Última actualización
- 30 oct 2022
- Guardar como PDF

( \newcommand{\kernel}{\mathrm{null}\,}\)
El foco de esta sección está en la emisión de radiación ultravioleta y visible tras la excitación térmica de los átomos. La espectroscopia de emisión atómica tiene una larga historia. Las aplicaciones cualitativas basadas en el color de las llamas se utilizaron en la fundición de minerales ya en 1550 y se desarrollaron más completamente alrededor de 1830 con la observación de los espectros atómicos generados por la emisión de llama y la emisión de chispas [Dawson, J. B. J. Anal. En. Espectrosc. 1991, 6, 93—98]. Las aplicaciones cuantitativas basadas en la emisión atómica de chispas eléctricas fueron desarrolladas por Lockyer a principios de 1870 y las aplicaciones cuantitativas basadas en la emisión de llama fueron pioneras por Lundegardh en 1930. La emisión atómica basada en la emisión de un plasma se introdujo en 1964.
Para una introducción en línea a gran parte del material de esta sección, consulte Espectroscopia de emisión atómica (AES) de Tomas Spudich y Alexander Scheeline, un recurso que forma parte de la Biblioteca Digital de Ciencias Analíticas.
Espectros de emisión atómica
La emisión atómica ocurre cuando un electrón de valencia en un orbital atómico de mayor energía regresa a un orbital atómico de menor energía. La Figura 10.7.1 muestra una porción del diagrama de niveles de energía para el sodio, que consiste en una serie de líneas discretas a longitudes de onda que corresponden a la diferencia de energía entre dos orbitales atómicos.
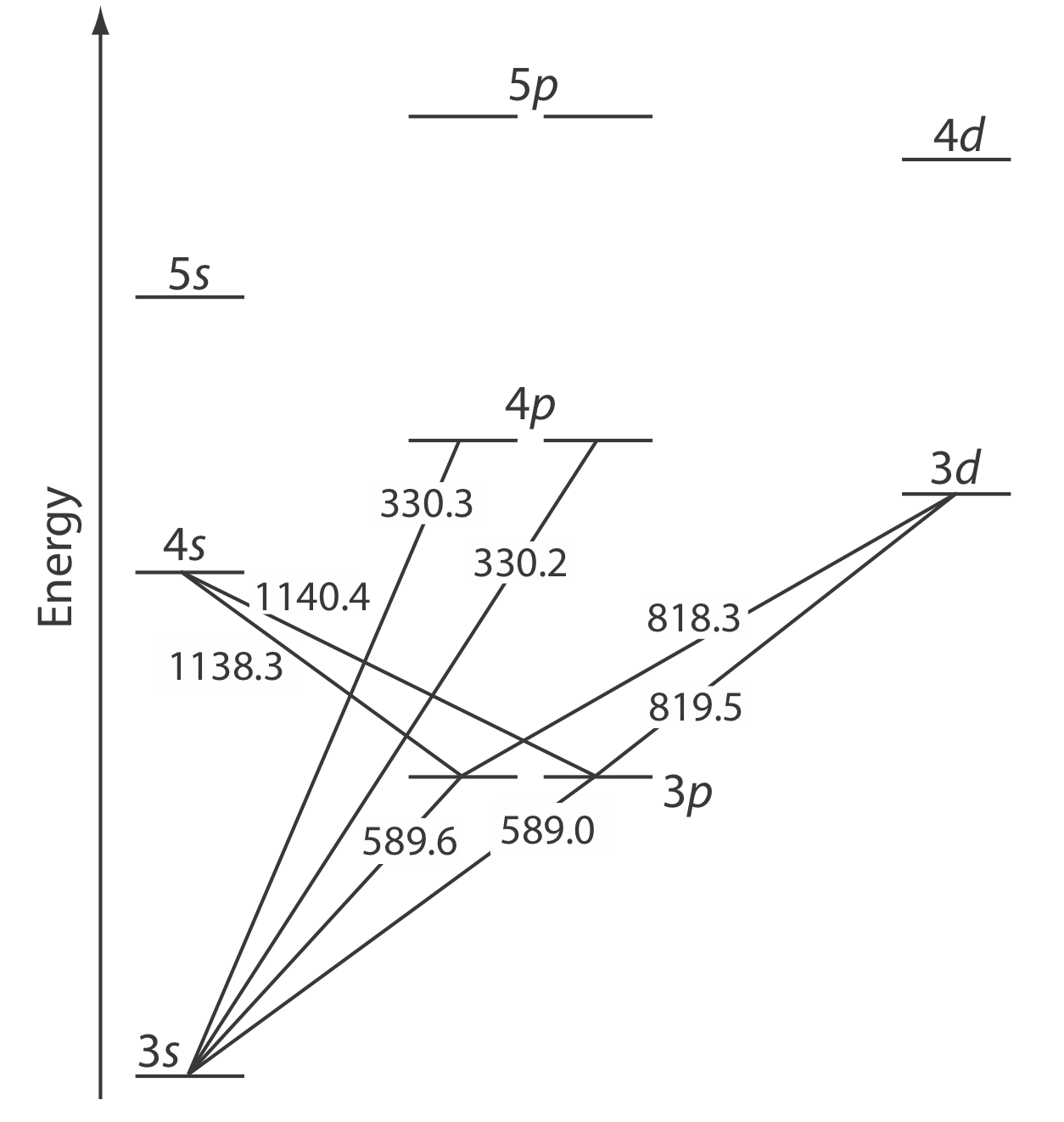
La intensidad de una línea de emisión atómica, I e, es proporcional al número de átomos,N∗, que pueblan el estado excitado,
donde k es una constante que da cuenta de la eficiencia de la transición. Si un sistema de átomos está en equilibrio térmico, la población de estado excitado i se relaciona con la concentración total de átomos, N, por la distribución de Boltzmann. Para muchos elementos a temperaturas inferiores a 5000 K la distribución de Boltzmann se aproxima como
donde g i y g 0 son factores estadísticos que dan cuenta del número de niveles de energía equivalentes para el estado excitado y el estado fundamental, E i es la energía del estado excitado en relación con un suelo energía del estado, E 0, k es la constante de Boltzmann (1.3807×10−23J/K), y T es la temperatura en Kelvin. De la Ecuación\ ref {10.2} esperamos que los estados excitados con energías más bajas tengan poblaciones más grandes y líneas de emisión más intensas. También esperamos que la intensidad de emisión aumente con la temperatura.
Equipo
Un espectrómetro de emisión atómica es similar en diseño a la instrumentación para absorción atómica. De hecho, es fácil adaptar la mayoría de los espectrómetros de absorción atómica de llama para la emisión atómica apagando la lámpara de cátodo hueco y monitoreando la diferencia entre la intensidad de emisión al aspirar la muestra y al aspirar un blanco. Sin embargo, muchos espectrómetros de emisión atómica son instrumentos dedicados diseñados para aprovechar características exclusivas de la emisión atómica, incluido el uso de plasmas, arcos, chispas y láseres como fuentes de atomización y excitación, y una capacidad mejorada para el análisis multielemental.
Atomización y Excitación
La emisión atómica requiere un medio para convertir en un átomo gaseoso libre un analito que esté presente en una muestra sólida, líquida o en solución. La misma fuente de energía térmica utilizada para la atomización suele servir como fuente de excitación. Los métodos más comunes son las llamas y los plasmas, los cuales son útiles para muestras líquidas o en solución. Las muestras sólidas se analizan disolviendo en un disolvente y usando un atomizador de llama o plasma.
Fuentes de llama
La atomización y excitación en la emisión atómica de llama se logra con el mismo conjunto de cámara de nebulización y pulverización utilizado en la absorción atómica (Figura 10.4.1). El cabezal del quemador consta de una o varias ranuras, o un quemador estilo Meker. Los instrumentos de emisión atómica más antiguos solían utilizar un quemador de consumo total en el que la muestra se extrae a través de un tubo capilar y se inyecta directamente en la llama.
Un quemador Meker es similar al quemador Bunsen más común que se encuentra en la mayoría de los laboratorios; está diseñado para permitir temperaturas más altas y una llama de mayor diámetro.
Fuentes de Plasma
Un plasma es un gas caliente parcialmente ionizado que contiene una abundante concentración de cationes y electrones. El plasma utilizado en la emisión atómica se forma ionizando una corriente que fluye de gas argón, produciendo iones de argón y electrones. La alta temperatura de un plasma resulta del calentamiento resistivo a medida que los electrones y los iones argón se mueven a través del gas. Debido a que un plasma opera a una temperatura mucho más alta que una llama, proporciona una mejor eficiencia de atomización y una mayor población de estados excitados.
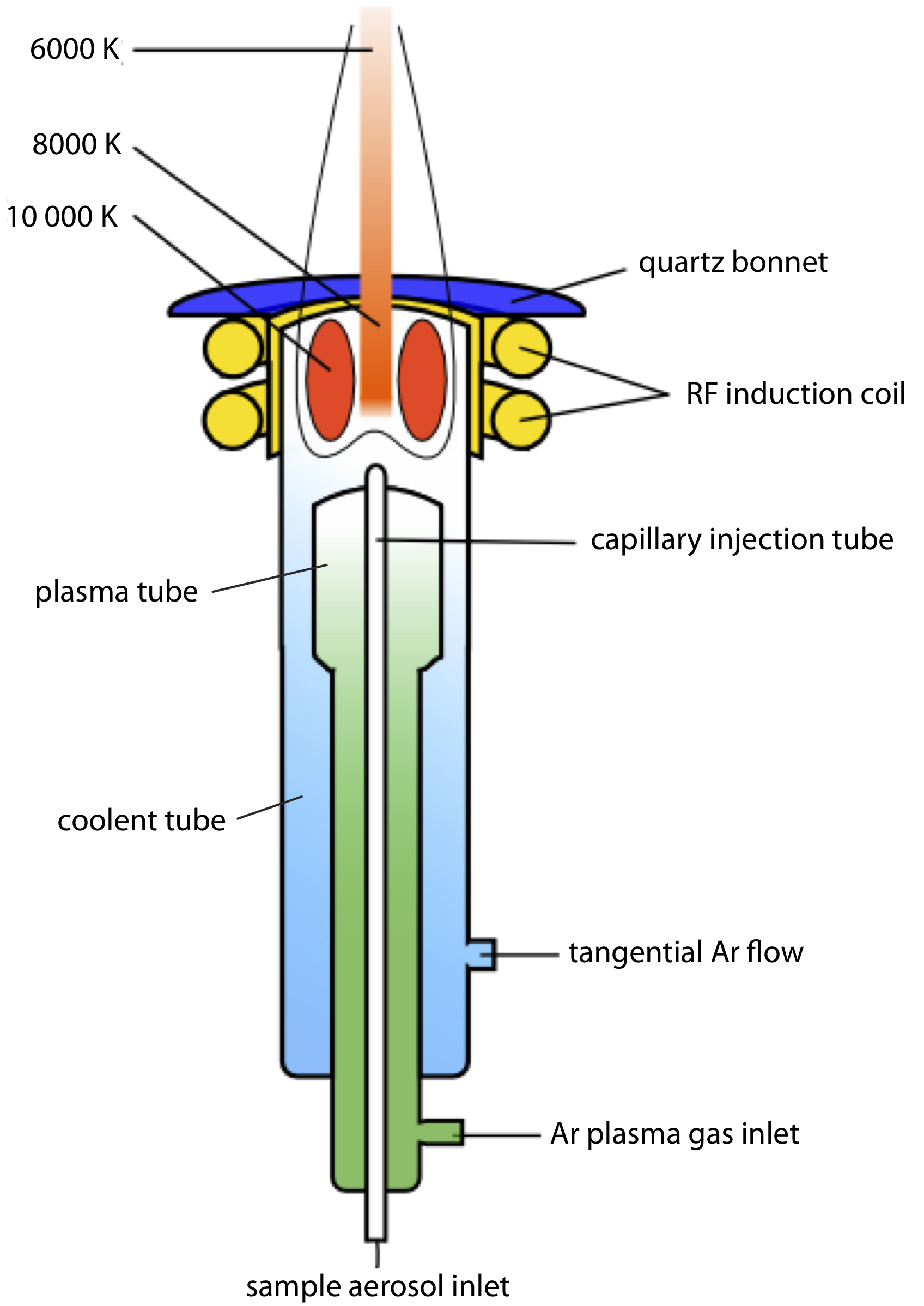
Un diagrama esquemático de la fuente de plasma acoplada inductivamente (ICP) se muestra en la Figura 10.7.2 . La antorcha ICP consta de tres tubos concéntricos de cuarzo, rodeados en la parte superior por una bobina de inducción de radiofrecuencia. La muestra se mezcla con una corriente de Ar usando un nebulizador, y se lleva al plasma a través del tubo capilar central de la antorcha. La formación de plasma es iniciada por una chispa de una bobina Tesla. Una corriente alterna de radiofrecuencia en la bobina de inducción crea un campo magnético fluctuante que induce a los iones argón y a los electrones a moverse en una trayectoria circular. Las colisiones resultantes con el abundante gas sindicalizado dan lugar a un calentamiento resistivo, proporcionando temperaturas tan altas como 10000 K en la base del plasma, y entre 6000 y 8000 K a una altura de 15—20 mm por encima de la bobina, donde generalmente se mide la emisión. A estas altas temperaturas el tubo exterior de cuarzo debe aislarse térmicamente del plasma. Esto se logra mediante el flujo tangencial de argón mostrado en el diagrama esquemático.
Análisis Multielemental
La espectroscopia de emisión atómica es ideal para un análisis multielemental ya que todos los analitos de una muestra se excitan simultáneamente. Si el instrumento incluye un monocromador de barrido, podemos programarlo para que se mueva rápidamente a la longitud de onda deseada de un analito, hacer una pausa para registrar su intensidad de emisión y luego pasar a la siguiente longitud de onda del analito. Este análisis secuencial permite una tasa de muestreo de 3—4 analitos por minuto.
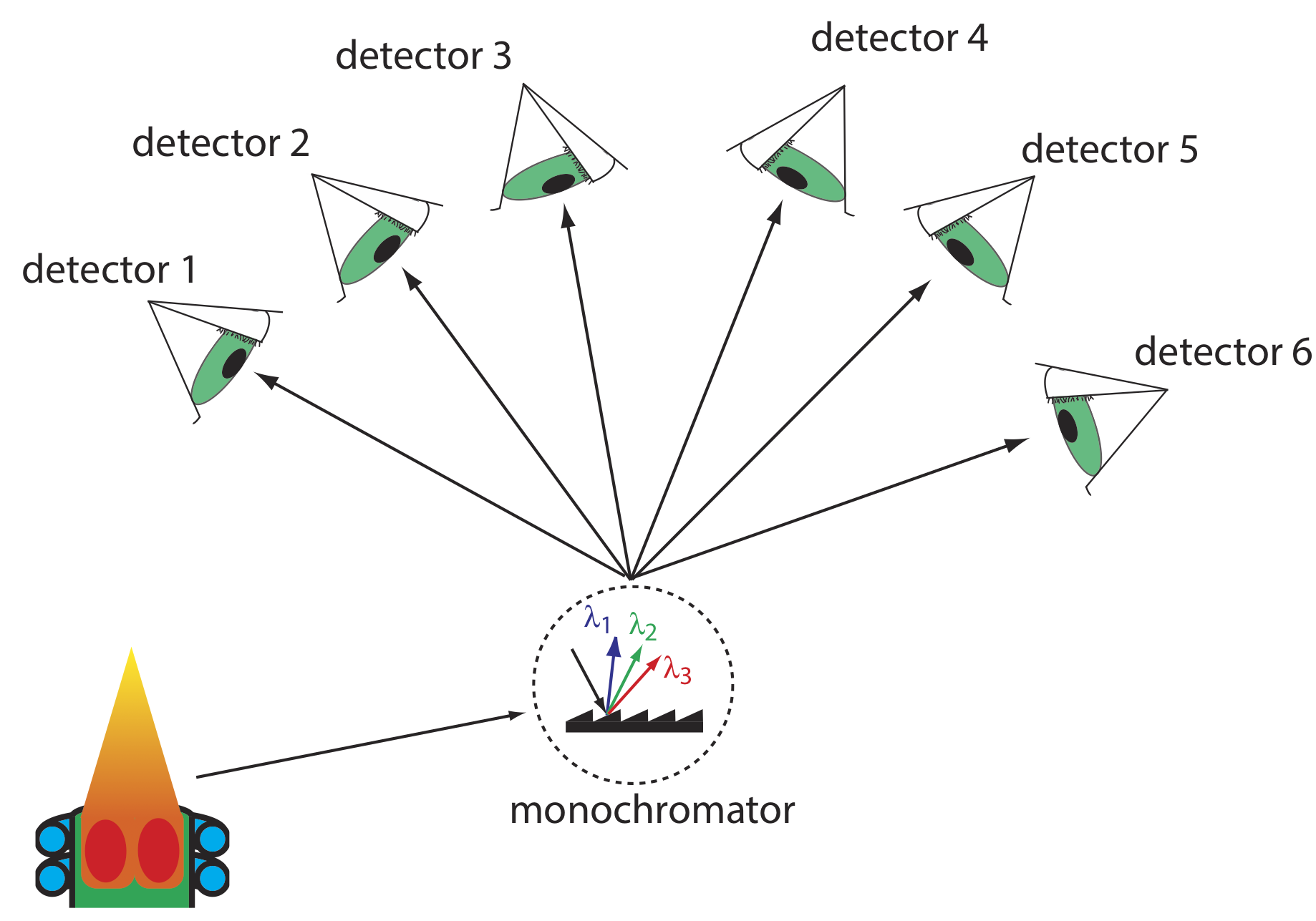
Otra aproximación a un análisis multielemental es utilizar un instrumento multicanal que nos permita monitorear simultáneamente muchos analitos. Un diseño simple para un espectrómetro multicanal, mostrado en la Figura 10.7.3 , conecta un monocromador con múltiples detectores que se colocan en una matriz semicircular alrededor del monocromador en posiciones que corresponden a las longitudes de onda de los analitos.
Aplicaciones Cuantitativas
La emisión atómica se usa ampliamente para el análisis de metales traza en una variedad de matrices de muestra. El desarrollo de un método cuantitativo de emisión atómica requiere varias consideraciones, entre ellas elegir una fuente para atomización y excitación, seleccionar una longitud de onda y ancho de hendidura, preparar la muestra para su análisis, minimizar las interferencias espectrales y químicas, y seleccionar un método de estandarización.
Elección de fuente de atomización y excitación
Excepto para los metales alcalinos, los límites de detección al usar un ICP son significativamente mejores que los obtenidos con emisión de llama (Cuadro 10.7.1 ). Los plasmas también están sujetos a menos interferencias espectrales y químicas. Por estas razones, una fuente de emisión de plasma suele ser la mejor opción.
Seleccionar la longitud de onda y el ancho de hendidura
La elección de la longitud de onda viene dictada por la necesidad de sensibilidad y la necesidad de evitar interferencias de las líneas de emisión de otros constituyentes en la muestra. Debido a que el espectro de emisión atómica de un analito tiene una abundancia de líneas de emisión, particularmente cuando se usa una fuente de plasma de alta temperatura, es inevitable que haya cierta superposición entre las líneas de emisión. Por ejemplo, un análisis para Ni usando la línea de emisión atómica a 349.30 nm se complica por la línea de emisión atómica para Fe a 349.06 nm.
Un ancho de hendidura más estrecho proporciona una mejor resolución, pero a costa de que menos radiación llegue al detector. El enfoque más fácil para seleccionar una longitud de onda es registrar el espectro de emisión de la muestra y buscar una línea de emisión que proporcione una señal intensa y se resuelva desde otras líneas de emisión.
Preparación de la Muestra
Las fuentes de llama y plasma son las más adecuadas para muestras en solución y en forma líquida. Si bien una muestra sólida puede analizarse insertándola directamente en la llama o plasma, generalmente se ponen primero en solución por digestión o extracción.
Minimizar interferencias espectrales
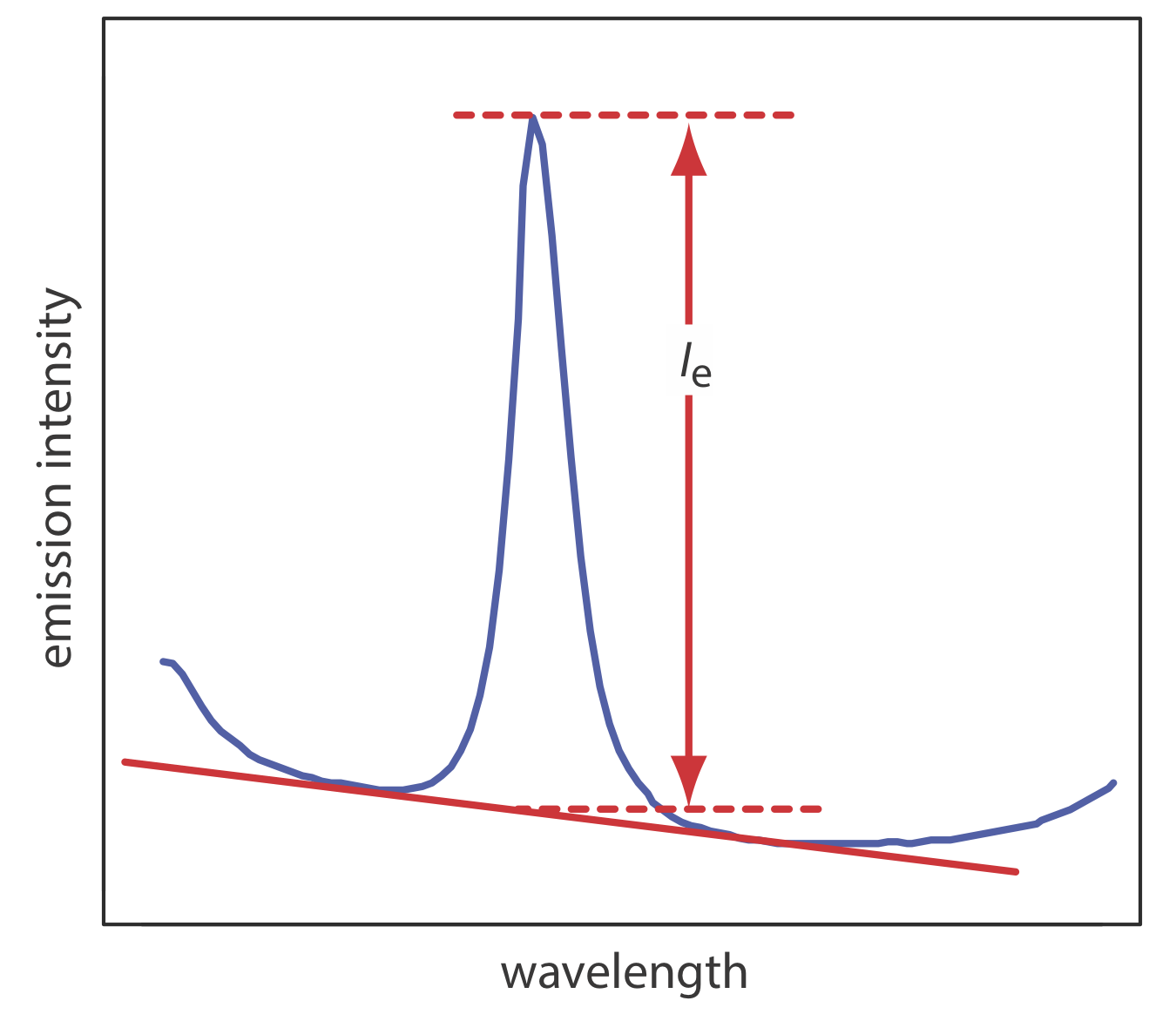
La interferencia espectral más importante es amplia, la emisión de fondo de la llama o plasma y las bandas de emisión de especies moleculares. Esta emisión de fondo es particularmente severa para las llamas debido a que la temperatura es insuficiente para descomponer compuestos refractarios, como óxidos e hidróxidos. Las correcciones de fondo para la emisión de llama se realizan escaneando sobre la línea de emisión y dibujando una línea base (Figura 10.7.4 ). Debido a que la temperatura de un plasma es mucho mayor, una interferencia de fondo debida a la emisión molecular es un problema menor. Aunque la emisión del núcleo del plasma es fuerte, es insignificante a una altura de 10—30 mm por encima del núcleo donde normalmente se realizan las mediciones.
Minimización de las interferencias químicas
La emisión de llama está sujeta a los mismos tipos de interferencias químicas que la absorción atómica; se minimizan usando los mismos métodos: ajustando la composición de la llama y agregando agentes protectores, agentes liberadores o supresores de ionización. Una interferencia química adicional es el resultado de la autoabsorción. Debido a que la temperatura de la llama es mayor en su centro, la concentración de átomos de analito en estado excitado es mayor en el centro de la llama que en sus bordes externos. Si un átomo de estado excitado en el centro de la llama emite un fotón, entonces un átomo de estado fundamental en el refrigerador, las regiones externas de la llama pueden absorber el fotón, lo que disminuye la intensidad de emisión. Para mayores concentraciones de analito la autoabsorción puede invertir el centro de la banda de emisión (Figura 10.7.5 ).
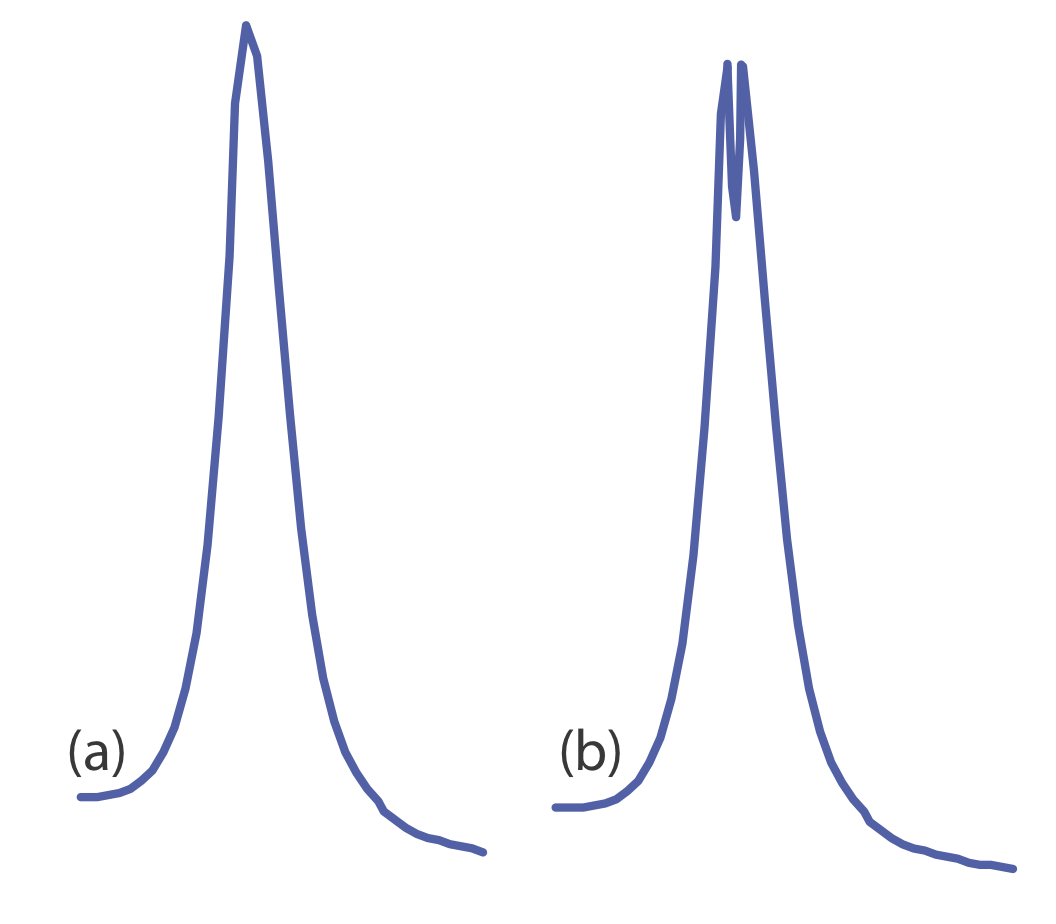
Las interferencias químicas al usar una fuente de plasma generalmente no son significativas debido a que la mayor temperatura del plasma limita la formación de especies no volátiles. Por ejemplo,PO3−4 es un interferente significativo al analizar muestras para Ca 2 + por emisión de llama, pero tiene un efecto insignificante al usar una fuente de plasma. Además, la alta concentración de electrones de la ionización del argón minimiza las interferencias de ionización.
Estandarización del Método
De la Ecuación\ ref {10.1} sabemos que la intensidad de emisión es proporcional a la población del estado excitado del analito,N∗. Si la llama o plasma está en equilibrio térmico, entonces la población en estado excitado es proporcional a la población total del analito, N, a través de la distribución de Boltzmann (Ecuación\ ref {10.2}).
Una curva de calibración para la emisión de llama generalmente es lineal en dos a tres órdenes de magnitud, con linealidad limitante de ionización cuando las concentraciones del analito son pequeñas y linealidad limitante de autoabsorción a concentraciones más altas de analito. Cuando se utiliza un plasma, que sufre menos interferencias químicas, la curva de calibración a menudo es lineal sobre cuatro a cinco órdenes de magnitud y no se ve afectada significativamente por los cambios en la matriz de los patrones.
La intensidad de emisión se ve afectada significativamente por muchos parámetros, incluyendo la temperatura de la fuente de excitación y la eficiencia de la atomización. Un incremento en la temperatura de 10 K, por ejemplo, produce un incremento de 4% en la fracción de átomos de Na en el estado excitado de 3 p, una incertidumbre en la señal que puede limitar el uso de estándares externos. El método de estándares internos se utiliza cuando las variaciones en los parámetros de origen son difíciles de controlar. Para compensar los cambios en la temperatura de la fuente de excitación, se selecciona el estándar interno para que su línea de emisión esté cerca de la línea de emisión del analito. Además, el estándar interno debe estar sujeto a las mismas interferencias químicas para compensar los cambios en la eficiencia de atomización. Para corregir con precisión estos errores, se monitorean simultáneamente las líneas de emisión de analitos y estándares internos.
Método Representativo 10.7.1: Determinación de Sodio en un Sustituto de Sal
La mejor manera de apreciar los detalles teóricos y prácticos discutidos en esta sección es examinar cuidadosamente un método analítico típico. Aunque cada método es único, la siguiente descripción de la determinación de sodio en sustitutos de sal proporciona un ejemplo instructivo de un procedimiento típico. La descripción aquí se basa en Goodney, D. E. J. Chem. Educ. 1982, 59, 875—876.
Descripción del método
Los sustitutos de sal, que se utilizan en lugar de la sal de mesa para individuos con dietas bajas en sodio, reemplazan el NaCl con KCl. Dependiendo de la marca, también están presentes ácido fumárico, hidrogenofosfato de calcio o tartrato de potasio. Aunque destinados a estar libres de sodio, los sustitutos de sal contienen pequeñas cantidades de NaCl como impureza. Por lo general, la concentración de sodio en un sustituto de sal es de aproximadamente 100 μg/g La concentración exacta de sodio se determina por emisión atómica de llama. Debido a que es difícil hacer coincidir la matriz de los estándares con la de la muestra, el análisis se realiza mediante el método de adiciones estándar.
Procedimiento
Se prepara una muestra colocando una porción de aproximadamente 10 g del sustituto de sal en 10 mL de HCl 3 M y 100 mL de agua destilada. Después de que la muestra se haya disuelto, se transfiere a un matraz aforado de 250 ml y se diluye a volumen con agua destilada. Se prepara una serie de adiciones estándar colocando porciones de 25 mL de la muestra diluida en matraces volumétricos separados de 50 mL, agregando a cada uno una cantidad conocida de una solución estándar de aproximadamente 10 mg/L de Na +, y diluyendo a volumen. Después de poner a cero el instrumento con un blanco apropiado, el instrumento se optimiza a una longitud de onda de 589.0 nm mientras se aspira una solución estándar de Na +. La intensidad de emisión se mide para cada una de las muestras estándar de adición y la concentración de sodio en el sustituto de la sal se reporta en μg/g.
Preguntas
1. El potasio se ioniza más fácilmente que el sodio. ¿Qué problema podría presentar esto si usa estándares externos preparados a partir de una solución madre de 10 mg Na/L en lugar de usar un conjunto de adiciones estándar?
Debido a que el potasio está presente a una concentración mucho mayor que la del sodio, su ionización suprime la ionización del sodio. Normalmente suprimir la ionización es algo bueno porque aumenta la intensidad de emisión. En este caso, sin embargo, la diferencia entre la matriz del patrón y la matriz de la muestra significa que el sodio en un estándar experimenta más ionización que una cantidad equivalente de sodio en una muestra. El resultado es un error determinado.
2. Una forma de evitar un error determinado al usar estándares externos es hacer coincidir la matriz de los estándares con la de la muestra. Podríamos, por ejemplo, preparar estándares externos usando KCl de grado reactivo para hacer coincidir la matriz con la de la muestra. ¿Por qué esto no es una buena idea para este análisis?
El sodio es un contaminante común en muchos productos químicos. KCl de grado reactivo, por ejemplo, puede contener 40—50 μg de Na/ g. Esta es una fuente significativa de sodio, dado que el sustituto de la sal contiene aproximadamente 100 μg Na/g.
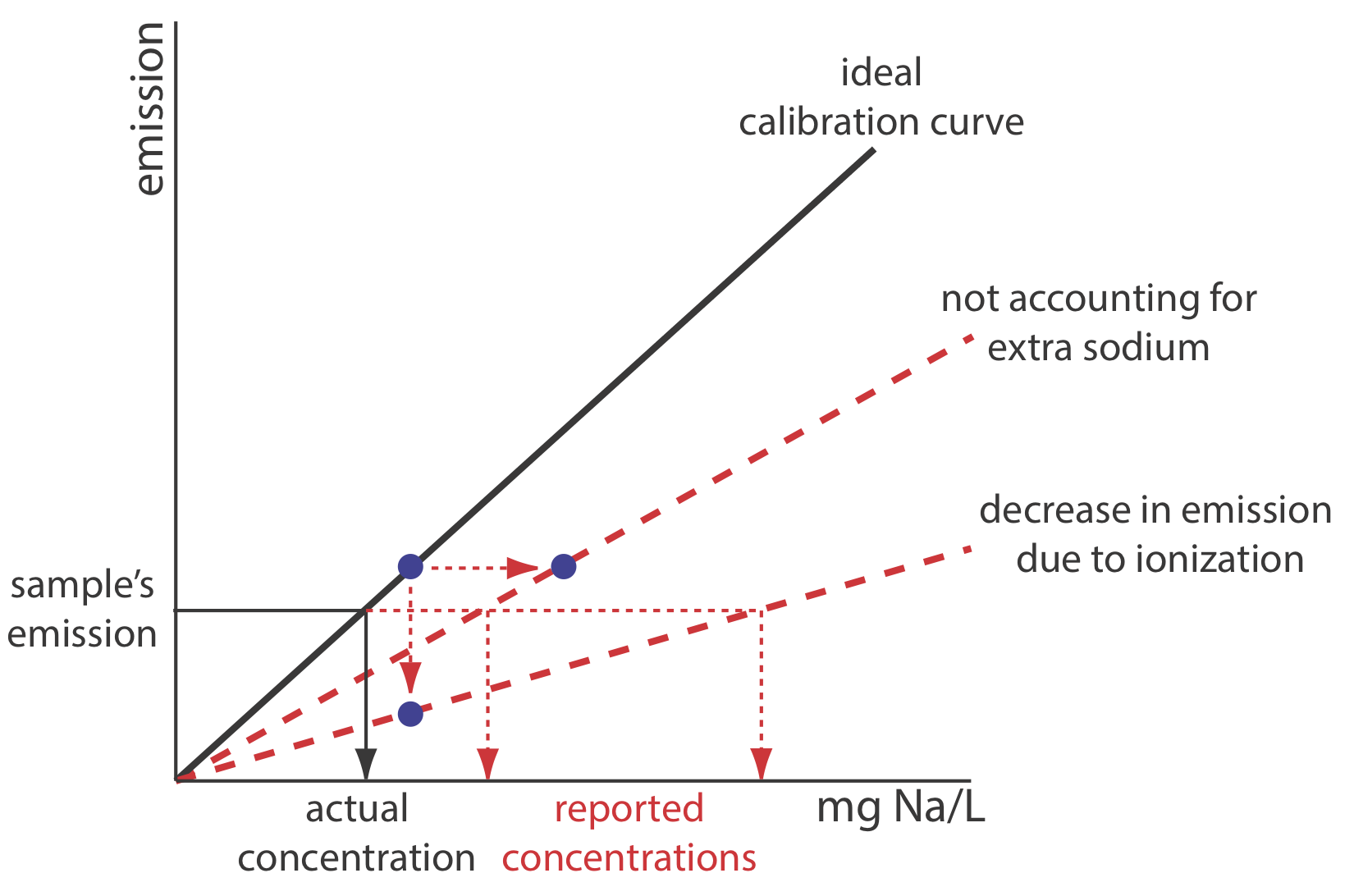
3. Supongamos que decide utilizar una estandarización externa. Dadas las preguntas anteriores, ¿es probable que el resultado de su análisis subestime o sobreestime la cantidad de sodio en el sustituto de la sal?
La línea negra continua en la Figura 10.7.6 muestra la curva de calibración ideal, asumiendo que coincidimos la matriz del estándar con la matriz de la muestra, y que lo hacemos sin agregar ningún sodio adicional. Si preparamos los estándares externos sin agregar KCl, la emisión para cada estándar disminuye debido al aumento de la ionización. Esto se muestra por la inferior de las dos líneas rojas discontinuas. La preparación de los estándares mediante la adición de KCl de grado reactivo aumenta la concentración de sodio debido a su contaminación. Debido a que subestimamos la concentración real de sodio en los estándares, la curva de calibración resultante se muestra por la otra línea roja discontinua. En ambos casos, la emisión de la muestra resulta en nuestra sobreestimación de la concentración de sodio en la muestra.
4. Un problema con el análisis de las muestras de sal es su tendencia a obstruir el conjunto del aspirador y quemador. ¿Qué efecto tiene esto en el análisis?
La obstrucción del conjunto aspirador y quemador disminuye la velocidad de aspiración, lo que disminuye la concentración del analito en la llama. El resultado es una disminución en la intensidad de emisión y un error negativo determinado.
Para evaluar el método descrito en el Método Representativo 10.7.1, se prepara una serie de adiciones estándar utilizando una muestra de 10.0077-g de un sustituto de sal. Aquí se muestran los resultados de un análisis de emisión atómica de llama de los estándares [Goodney, D. E. J. Chem. Educ. 1982, 59, 875—876].
| Na añadido (µg/mL) | I e (unidades arb.) |
|---|---|
| 0.000 | 1.79 |
| 0.420 | 2.63 |
| 1.051 | 3.54 |
| 2.102 | 4.94 |
| 3.153 | 6.18 |
Cuál es la concentración de sodio, en μg/g, en el sustituto de la sal.
Solución
La regresión lineal de la intensidad de emisión versus la concentración de Na agregado da la curva de calibración de adiciones estándar que se muestra a continuación, la cual tiene la siguiente ecuación de calibración.
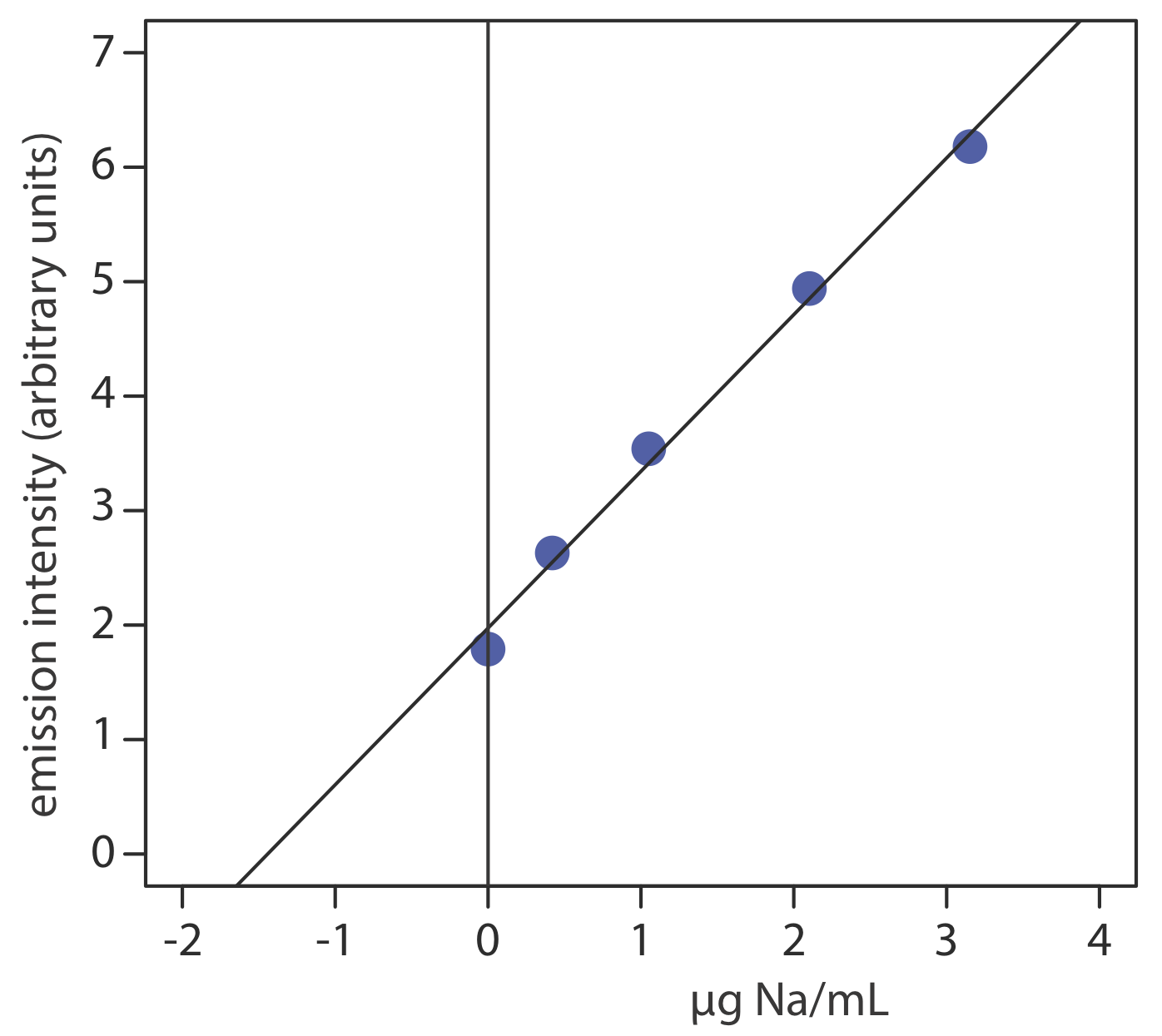
Ie=1.97+1.37×μg NamL
La concentración de sodio en la muestra es el valor absoluto de la intersección x de la curva de calibración. Sustituir cero por la intensidad de emisión y resolver la concentración de sodio da un resultado de 1.44 μGNA/ml. La concentración de sodio en el sustituto de la sal es
1.44 μg NamL×50.00 mL25.00 mL×250.0 mL10.0077 g sample =71.9 μg Na/g
Evaluación de Espectroscopia de Emisión Atómica
Escala de Operación
La escala de operaciones para la emisión atómica es ideal para el análisis directo de analitos traza y ultratraza en muestras macro y meso. Con diluciones apropiadas, la emisión atómica se puede aplicar a analitos mayores y menores.
Precisión
Cuando las interferencias espectrales y químicas son insignificantes, la emisión atómica puede lograr resultados cuantitativos con precisiones del 1— 5%. Para la emisión de llama, la precisión con frecuencia está limitada por interferencias químicas. Debido a que la mayor temperatura de una fuente de plasma da lugar a más líneas de emisión, la precisión al usar la emisión de plasma a menudo está limitada por la radiación parásita de las líneas de emisión superpuestas.
Precisión
Para muestras y estándares en los que la concentración del analito excede el límite de detección en al menos un factor de 50, la desviación estándar relativa para la emisión de llama y plasma es de aproximadamente 1— 5%. Quizás el factor más importante que afecta a la precisión es la estabilidad de la temperatura de la llama o del plasma. Por ejemplo, en una llama de 2500 K una fluctuación de temperatura de±2.5 K da una desviación estándar relativa de 1% en la intensidad de emisión. Se logran mejoras significativas en la precisión al usar estándares internos.
Sensibilidad
La sensibilidad está influenciada por la temperatura de la fuente de excitación y la composición de la matriz de muestra. La sensibilidad se optimiza aspirando una solución estándar de analito y maximizando la emisión ajustando la composición de la llama y la altura desde la cual monitoreamos la emisión. Las interferencias químicas, cuando están presentes, disminuyen la sensibilidad del análisis. Debido a que la sensibilidad de la emisión plasmática se ve menos afectada por la matriz de muestra, es posible una curva de calibración preparada usando estándares en una matriz de agua destilada incluso para muestras que tienen matrices más complejas.
Selectividad
La selectividad de la emisión atómica es similar a la de la absorción atómica. La emisión atómica tiene la ventaja adicional de un análisis rápido secuencial o simultáneo de múltiples analitos.
Tiempo, Costo y Equipo
El rendimiento de la muestra con emisión atómica es rápido cuando se utiliza un sistema automatizado que puede analizar múltiples analitos. Por ejemplo, son posibles tasas de muestreo de 3000 determinaciones por hora usando un ICP multicanal, y tasas de muestreo de 300 determinaciones por hora cuando se usa un ICP secuencial. La emisión de llama a menudo se logra usando un espectrómetro de absorción atómica, que generalmente cuesta entre $10,000 y $50,000. Los ICP secuenciales varían en precio de $55,000—$150,000, mientras que un ICP capaz de análisis multielemental simultáneo cuesta entre $80,000—$200,000. Los ICP combinados que son capaces de realizar análisis secuenciales y simultáneos varían en precio de $150,000 a $300,000. El costo de Ar, que se consume en cantidades significativas, no puede pasarse por alto al considerar el gasto de operar un ICP.