
8.4: Electrofilos




- Última actualización
- 30 oct 2022
- Guardar como PDF

( \newcommand{\kernel}{\mathrm{null}\,}\)
A continuación, pasamos a los electrófilos. En la gran mayoría de las reacciones de sustitución nucleofílica verá en este y otros textos de química orgánica, el átomo electrófilo es un carbono unido a un átomo electronegativo, generalmente oxígeno, nitrógeno, azufre, o un halógeno. El concepto de electrofilicidad es relativamente simple: un átomo pobre en electrones es un objetivo atractivo para algo que es rico en electrones, es decir, un nucleófilo. Sin embargo, también debemos considerar el efecto del impedimento estérico sobre la electrofilicidad.
Obstén estérico en el electrófilo
Uno de los factores más importantes a considerar al observar el electrófilo en una reacción de sustitución nucleofílica es el impedimento estérico. Consideremos dosSN2 reacciones hipotéticas: una en la que el electrófilo es un carbono metílico y otra en la que es carbono terciario.
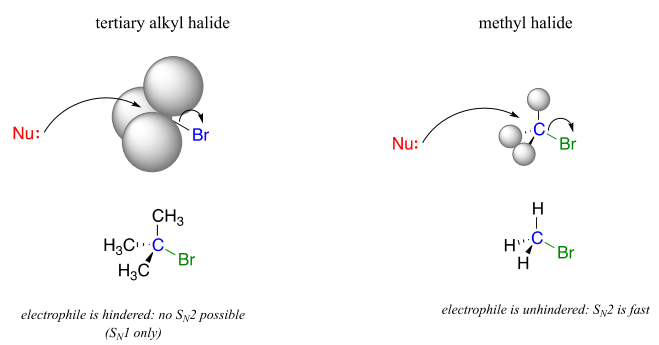
Debido a que los tres sustituyentes en el electrófilo de metilo son átomos de hidrógeno, el nucleófilo tiene un camino relativamente claro para el ataque posterior, y laSN2 reacción tendrá lugar fácilmente. Sin embargo, el ataque posterior al electrófilo de carbono terciario está bloqueado por los grupos metilo voluminosos, impidiendo el acceso al sitio de electrofilicidad.
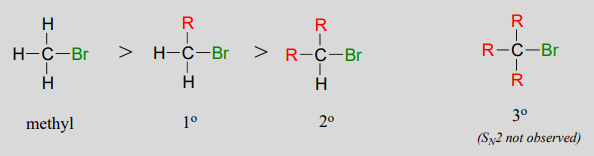
SN2las reacciones ocurren en electrófilos metílicos, primarios y secundarios de carbono. El grado de impedimento estérico determina las velocidades relativas de reacción: los electrófilos de metilo sin obstáculos reaccionan más rápido y los electrófilos secundarios de carbono más obstaculizados reaccionan más lentamente, asumiendo que todas las demás condiciones de reacción son idénticas. SN2las reacciones no ocurren en un grado apreciable en electrófilos de carbono terciario.
¿Cuál se esperaría que reaccionara más rápidamente en unaSN2 reacción con un ion azida (N−3) nucleófilo en acetona solvente: 1-bromo-2,2-dimetilbutano o 1-bromo-3-metilbutano?
¿Qué pasa con elSN1 camino? El impedimento estérico alrededor del carbono electrófilo no es un factor significativo para ralentizar unaSN1 reacción. Esto tiene perfecto sentido desde un punto de vista geométrico: las limitaciones impuestas por los estéricos son significativas en unSN2 desplazamiento porque el electrófilo que se está atacando es un carbono tetraédrico hibridado sp3 con ángulos relativamente 'apretados' de109.5∘. Recuerde que en unSN1 mecanismo, el grupo de salida sale primero, y luego el nucleófilo ataca a un intermedio de carbocatiónsp2 -hibridado, que tiene geometría plana trigonal con120∘ ángulos 'abiertos'.
Se necesitan obras de arte
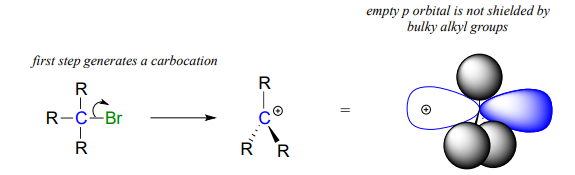
Con esta geometría abierta, el orbital p vacío del carbocatión ya no está significativamente protegido del nucleófilo que se aproxima por los grupos alquilo voluminosos, y es un 'objetivo fácil' para un nucleófilo: este paso es rápido, y no es el paso determinante de la velocidad para unaSN1 reacción.
Estabilidad de carbocationes
¿Cuáles son, entonces, las características de un electrófilo que favorecen una vía deSN1 reacción en lugar de unaSN2 vía? Sabemos que el paso limitante de velocidad de unaSN1 reacción es el primer paso: pérdida del grupo lábil y formación del intermedio carbocatión. Por consiguiente, la velocidad de unaSN1 reacción depende en gran medida de la estabilidad del intermedio de carbocatión.
La pregunta crítica ahora se convierte en:
¿Qué estabiliza un carbocatión?
Piense en el Capítulo 7, cuando estábamos aprendiendo a evaluar la fuerza de un ácido. La pregunta crítica que hubo fue: “¿qué tan estable es la base conjugada que resulta cuando este ácido dona su protón”? En muchos casos, esta base conjugada era un anión, un centro de exceso de densidad electrónica. Cualquier cosa que pueda alejar parte de esta densidad de electrones —en otras palabras, cualquier grupo aceptor de electrones— estabilizará el anión.
Por el contrario, un carbocatión es estabilizado por un grupo donador de electrones, y de estabilizado por un grupo aceptor de electrones.
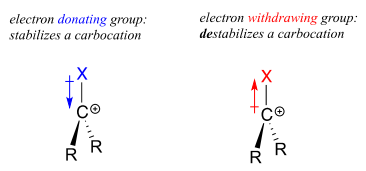
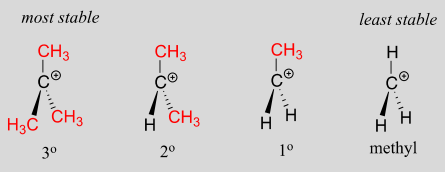
Una especie cargada positivamente como un carbocatión es pobre en electrones y, por lo tanto, cualquier cosa que dona densidad de electrones al centro de la pobreza electrónica ayudará a estabilizarla. Los grupos alquilo, debido a los electrones en sus enlaces carbono-carbono y carbono-hidrógeno, son grupos donadores de electrones débiles y estabilizarán los carbocationes cercanos. Lo que esto significa es que, en general, más carbocationes sustituidos son más estables: un carbocatión terc-butilo, por ejemplo, es más estable que un carbocatión isopropilo. Los carbocationes primarios son altamente inestables y no suelen observarse como intermedios de reacción; los cationes metilo son aún menos estables.
Más carbocationes sustituidos son más estables:
Otra forma de explicar esta tendencia en la estabilidad de los carbocationes implica el fenómeno de la hiperconjugación, en el que elp orbital vacío de un carbocatión se estabiliza por solapamiento con unσ enlace sobre un carbono adyacente. Esta superposición propaga efectivamente la carga positiva sobre un área más grande. La siguiente figura muestra elp orbital vacío de un carbocatión secundario siendo estabilizado por hiperconjugación con unC−Hσ enlace adyacente.
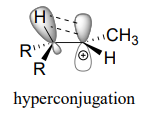
La hiperconjugación no es posible con un catión metilo ya que no hay ningúnσ enlace adyacente disponible para superponerse alp orbital vacío. A medida que aumenta el grado de sustitución en un carbocatión, también lo hace la capacidad de estabilizar las interacciones de hiperconjugación.
La presencia de un grupo aceptor de electrones -como un átomo de flúor- desestabilizará significativamente un carbocatión a través del efecto inductivo.

Los grupos carbonilo son extractores de electrones por efectos inductivos, debido a la polaridad delC=O doble enlace. Es posible demostrar en el laboratorio (veremos cómo en el problema 14.x) que el carbocatión A a continuación es más estable que el carbocatión B, aunque A es un carbocatión primario y B es secundario.
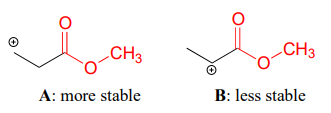
La carga positiva en el catión B está más cerca de la sustitución de carbonilo aceptor de electrones, y como aprendimos en la sección 7.3, el efecto inductivo de un grupo aceptor de electrones disminuye con la distancia.
La estabilización de un carbocatión también puede ocurrir a través de efectos de resonancia. Recordemos de la sección 7.4 que la carga negativa sobre un ion fenolato se estabiliza por resonancia, ya que la carga puede deslocalizarse a tres de los carbonos del anillo aromático.
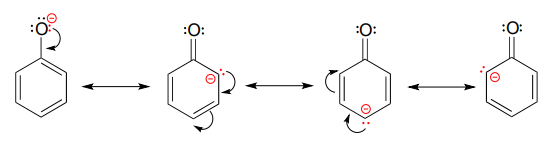
Una carga positiva también se estabiliza cuando puede deslocalizarse sobre más de un átomo. Considere un carbocatión bencílico, donde el carbono cargado positivamente está unido directamente a un anillo aromático. Un carbocatión bencílico se estabiliza por el efecto donador de electrones de resonancia del anillo aromático. Se pueden dibujar tres estructuras de resonancia adicionales para el carbocatión en el que la carga positiva se encuentra en uno de tres carbonos aromáticos:
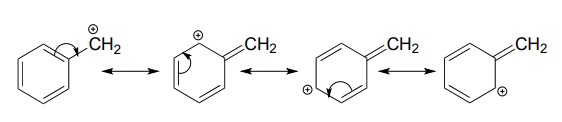
Rellene los números que faltan en esta declaración: El\(p\) sistema conjugado en el carbocatión bencílico anterior está compuesto por ______\(p\) orbitales que se superponen para compartir ______\(\pi \) electrones.
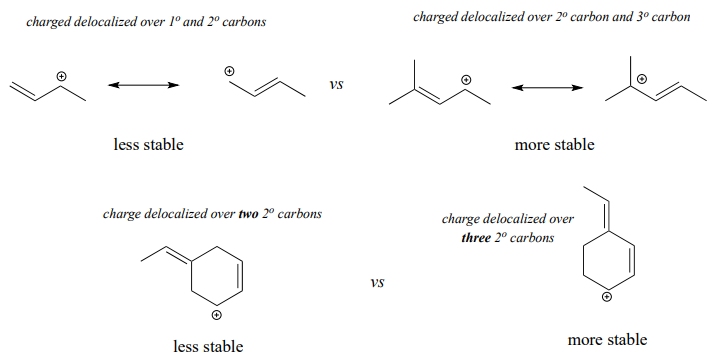
Los carbocationes alílicos, donde el carbono cargado positivamente es adyacente a un doble enlace, se estabilizan por deslocalización por resonancia de la carga positiva.
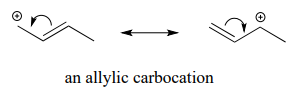
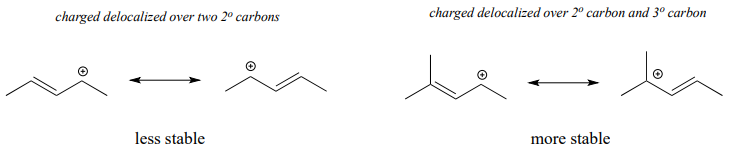
A menudo, debemos considerar más de un factor a la hora de predecir la estabilidad de los carbocationes. Por ejemplo, el carbocatión de la derecha en la figura siguiente es más estable que el carbocatión de la izquierda. Ambos son alílicos con la carga deslocalizada sobre dos carbonos, pero en el carbocatión más estable, uno de los carbonos es terciario.
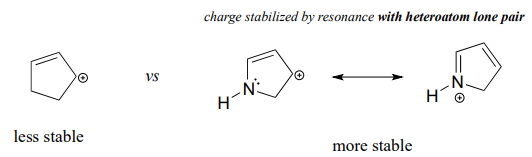
Debido a que los heteroátomos como el oxígeno y el nitrógeno son más electronegativos que el carbono, podría esperarse que serían grupos extractores de electrones desestabilizantes de carbocationes. De hecho, lo contrario suele ser cierto: si el átomo de oxígeno o nitrógeno está en la posición correcta, el efecto general puede ser la estabilización de carbocationes. Si bien estos heteroátomos son en efecto grupos extractores de electrones por inducción, pueden ser grupos donadores de electrones por resonancia y, como aprendimos anteriormente (sección 7.3) en el contexto de la química ácido-base, los efectos de resonancia son en general más potentes que los efectos inductivos cuando los dos operan en opuestos direcciones.
Considere los dos pares de especies de carbocationes a continuación:
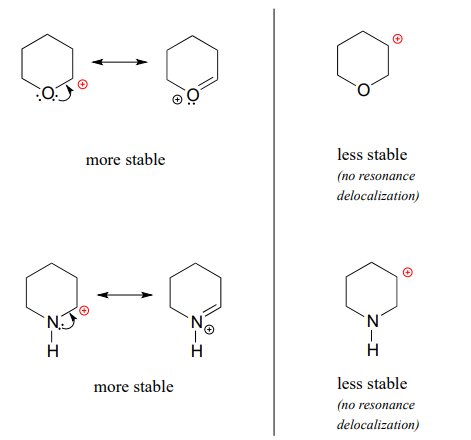
En los carbocationes más estables, el heteroátomo actúa como un grupo donador de electrones por resonancia: en efecto, el par solitario en el heteroátomo está disponible para deslocalizar la carga positiva. Obsérvese también que cada átomo en el contribuyente de resonancia principal tiene un octeto completo de electrones de valencia.
Se incluyen los siguientes carbocationes de más a menos estables:
Finalmente, los carbocationes vinílicos, en los que la carga positiva reside en un carbono de doble enlace, son altamente inestables.
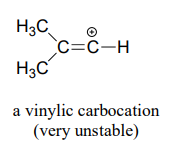
Explique por qué los carbocationes vinílicos son inestables. (Pista:)
- Insinuación
-
T hink sobre hibridación y electronegatividad.
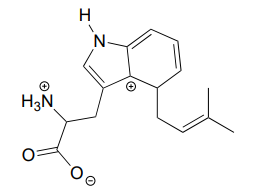
El carbocatión a continuación es una especie intermedia en una reacción que forma parte de la biosíntesis de un compuesto alucinógeno en un hongo. Dibujar un contribuyente de resonancia que muestre cómo se estabiliza por resonancia con el átomo de nitrógeno.
En su mayor parte, los carbocationes -incluso los carbocationes “relativamente estables” como los terciarios y/o bencílicos- siguen siendo especies intermedias transitorias altamente reactivas en reacciones orgánicas, que se forman brevemente y luego vuelven a reaccionar de inmediato. Sin embargo, hay algunos ejemplos inusuales de especies de carbocationes que son tan estables que pueden colocarse en un frasco y almacenarse en la repisa como sal. Violeta cristalina es el nombre común para la sal de cloruro del carbocatión cuya estructura se muestra a continuación. Observe las posibilidades estructurales para una amplia deslocalización por resonancia de la carga positiva y la presencia de tres grupos amina donadores de electrones.
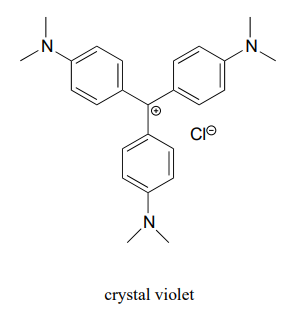
- Dibujar una estructura de resonancia del catión cristal violeta en la que la carga positiva se deslocaliza en uno de los átomos de nitrógeno.
- Observe que el violeta cristal está profundamente coloreado. Explica por qué podrías haber predicho esto observando su estructura química.
- El sistema conjugado de cristal violeta consiste en ¿cuántos orbitales p superpuestos que comparten cuántos electrones p?
- Los carbocationes más sustituidos son más estables que los carbocationes menos sustituidos (por ejemplo, los carbocationes terciarios son más estables que los secundarios).
- Los átomos electronegativos cercanos pueden disminuir la estabilidad de carbocationes por el efecto inductivo.
- Los carbocationes alílicos y bencílicos se estabilizan por deslocalización por resonancia de la carga positiva.
- La deslocalización de la carga positiva por resonancia con los electrones de par solitario en un heteroátomo contribuye a la estabilidad de los carbocationes.
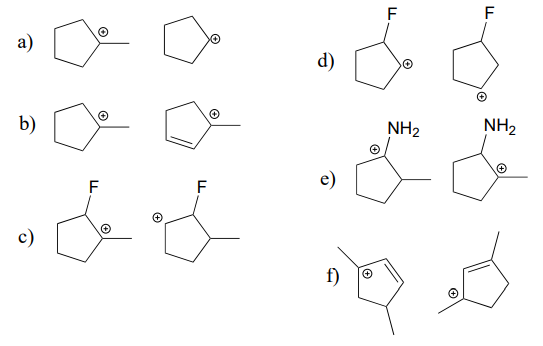
A continuación hay tres ejemplos que ilustran cómo podemos hacer predicciones sobre la estabilidad relativa de los carbocationes:
Afirma qué carbocatión en cada par a continuación es más estable, o si se espera que sean aproximadamente iguales. Explica tu razonamiento.
Ahora, volvamos a nuestra discusión sobre el electrófilo en unaSN1 reacción:
UnaSN1 reacción requiere un intermedio de carbocatión estabilizado. Cuanto más estable sea el carbocatión intermedio relevante, más favoreció la vía deSN1 reacción.
SN1las reacciones en general no ocurren en electrófilos de metilo o carbono primario: los intermedios de carbocationes involucrados serían demasiado inestables y la etapa determinante de la velocidad (generación de carbocationes) tendría una barrera energética muy alta. La sustitución en estos electrófilos ocurrirá a través de laSN2 vía.
La vía deSN1 reacción es posible, sin embargo, con electrófilos secundarios y terciarios de carbono, o con cualquier otro electrófilo de carbono en el que la salida del grupo lápida genere un carbocatión que se estabiliza por resonancia.
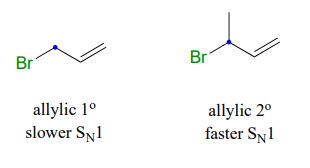
Por ejemplo: no se esperaría que un bromuro de alquilo primario sufriera sustitución nucleofílica por laSN1 vía. Un bromuro alílico de alquilo primario, por otro lado, generaría un carbocatión alílico relativamente estable y así laSN1 ruta es posible.

Un bromuro alílico de alquilo secundario sufriríaSN1 sustitución más rápidamente que el bromuro alílico de alquilo primario, debido a que el carbocatión relevante está más sustituido y por lo tanto más estable.
sp2-carbones hibridizados
La sustitución nucleofílica generalmente no ocurre en los carbonossp2 hibridados, ya sea por laSN1 víaSN2 o por la vía.
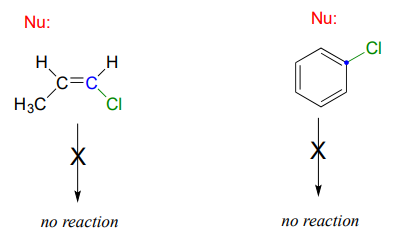
Los enlaces en los carbonossp2 hibridados son inherentemente más cortos y más fuertes que los enlaces en los carbonossp3 hibridados, lo que significa que es más difícil romper el enlace entre unsp2 carbono y un grupo potencial de salida (como el átomo de cloro en la figura anterior). Además, las consideraciones estéricas juegan un papel aquí: para atacar desde atrás al grupo de salida de una maneraSN2 similar, el nucleófilo tendría que acercarse en el plano del doble enlace carbono-carbono.
La sustitución por unaSN1 vía es igualmente improbable debido a la inestabilidad inherente de un carbocatión vinílico (doble enlace).